- 이 중 GalNAc conjugate 방법은 최근 FDA 승인된 RNAi 치료제 Givosiran에 적용된 기술로 delivery system 중 완성된 기술로 평가받고 있음.4 현재 GalNAc conjugates 방법은 siRNA 치료제 임상시험에서 5건의 후기임상(임상3상 및 등록심사 단계), 10건의 초기 임상시험(임 상1-2상)에 적용되고 있음.4 [그림 1] GalNAc-siRNA conjugates 작용 기전4 4 RNAi 치료제 개발 동향 RNAi 치료제 임상시험 - 2020년 11월 기준 임상시험 중인 RNAi 치료제는 총 51건으로 나타남.5-7 이 중 승인 건수를 포함한 임상3상 9건, 임상2상 13건, 확인되지 않는 임상현황을 포함한 임상1상 29건으로 확 인됨. 적응증별로 구별하였을 때 내분비계질환을 포함한 심혈관계질환이 15건, 암 13건, 감염 질환 8건, 안과질환 5건, 기타질환 10건으로 분류됨. Alnylam Pharmaceuticals가 가장 많은 10개의 파이프라인을 보유하고 있는 것으로 나타났으며, 이 중 2건은 FDA 품목 승인을 받음. 국내 기업의 경우 Hugel, Olix가 각 1건씩 siRNA 치료제 임상시험 중임(표 2).
siRNA는 RNAi 현상에 중요한 역할을 담당하고 있지만 임 상적으로 적용하기에는 여전히 장벽들이 존재한다 (그림 2). 첫 번째로, siRNA는 생체 내에서 불안정하다. 정맥주사를 한 경우 siRNA의 50%가 30분 이내에 사라진다. 그 이유로는 혈 중에서 효소에 의해 쉽게 분해 되고 작은 사이즈로 인해 신장 에서 빠르게 제거 된다. 몇몇 연구에서도 siRNA가 정맥 투여 시 신장에서 가장 높게 분포한다고 보고된다 [3]. 또한 가장 큰 장벽으로는 RNAi 나노 입자들이 혈액 순환시 간, 췌장에 서 옵소닌 작용에 의해 RES (Reticuloendothelial system)/ MPS (mononuclear phagocytic system)에 인식되어 쉽게 제거 되므로 치료효과가 감소한다 [4-5]. 두 번째는, 표면 이 음전하 (40-50 negative phosphate charge)를 띄고 있 기 때문에 siRNA 자체만으로는 쉽게 세포막을 통과하기 어 렵다. 따라서 siRNA를 세포 안으로 전달하기 위해서는 전달 체 개발이 요구된다. 또한 세포내로 전달이 되더라도 엔도좀 밖으로 방출되는데 어려움이 있을 수 있다. 세 번째는, 체내 로 들어간 siRNA가 표적하고 있지 않은 mRNA를 인식하는 off-target-effect로 원하지 않는 단백질 합성을 저해할 수 있다 [6]. 마지막으로, siRNA를 고농도로 처리할 경우 면역 반응을 유도하여 부작용을 초래한다. 따라서 siRNA를 매개 로 발생하는 면역반응에 대해 더 많은 연구들이 선행 되어야 한다.
siRNA 효율적으로 전달하기 위해서는 앞서 서술한 물 리적 장벽들을 해결해야 한다.
우선, siRNA의 구조적 변 형을 통해 혈중 내 안정성 및 전달 효율을 높인다. 주로 많 이 사용하는 방법으로 당의 2’-OH를 2-O-methyl, 2-H, 2-fluoro와 같은 다른 화학적 그룹들로 치환해준다 [7]. 이 렇게 변형된 구조들은 효소에 덜 영향을 받고 열에도 안정 적인 구조를 갖게 된다 [8]. 또한 센스가닥의 5’부분의 구 조적 변형과 안티센스 가닥의 3’ 말단에 overhang 구조 를 적용하여 표적 하는 mRNA의 특이성을 증가 시켜 offtarget effect를 줄일 수 있다 [9-10] (그림 3 a).
다음은 Naked siRNA를 세포 내로 전달을 용이하기 위해 다양한 나노 입자들의 전달체 개발이 요구된다. 다양한 양이온 전 달체를 이용하여 siRNA와 복합체를 형성해 전달 및 치료 효과를 높일 수 있다. 양이온 물질들은 주로 리포좀, PEI (polyethylenimine), PLL (poly-l-lysine), 키토산, 덴드리 머와 같은 폴리머를 사용하고 있다 (그림 3 b, c).
양이온 폴 리머는 물에 잘 용해 되고, 매우 높은 양전하를 갖고 있어서 쉽게 siRNA와 결합할 수 있는 장점이 있다. 또한 혈중에 분 포된 다양한 RNase로부터 보호해 naked siRNA의 분해를 감소시켜 siRNA의 전달효율이 높아질 수 있다. 아미노 그 룹이 풍부한 PEI 합성 폴리머가 siRNA 전달에 많이 이용되 고 있다. 세포 실험에서는 전달효율이 매우 높지만, 정맥주 사 후에는 옵소닌과 결합해 낮은 전달 효과와 독성을 나타내 는 단점을 갖고 있다.
덴드리머는 가지구조를 가지고 있어서 사이즈, 구조를 일정하게 조절할 수 있고, 전하를 가지고 있 어 siRNA전달에 용이하다. 그 종류로는 poly amidoamine (PAMAM)과 poly (propylenimine) (PPI) 등이 있고 표면 에 아민 그룹을 갖고 있어서 복합체 형성시 RNase 효소로 부터 보호하여 분해를 막고 전달 효율을 향상 시켜준다. 덴 드리머 안쪽의 아민 그룹은 프로톤 스폰지 효과에 의해 세 포 내로 들어갔을 때 엔도솜에서 siRNA가 잘 방출 될 수 있도록 도와주는 역할을 하고 있다 [11].
키토산 폴리머는 D-glucoseamines 과 N-acetyl-D-glucosamines로 구 성 되어있고, acetyl-D-glucosamines의 일차 아민이 pH 6.2-7.0 상태에서 siRNA와 결합하여 복합체를 형성한다. 키토산은 낮은 독성 및 면역반응을 나타내므로 생체에 적합 한 물질로 작용한다.
키토산/siRNA 복합체는 CHOK1 과 HEK 293 세포에서 높은 전달 효율을 보여준다 [12]. 하지 만 키토산/siRNA 복합체는 수용액 상태에서 매우 낮은 용해 성을 갖고 있기 때문에 in vivo 상태에서는 낮은 전달효율을 나타낸다 [13]. 이러한 문제점을 해결하기 위해, 키토산 표 면에 polyarginine과 PEG를 결합해 혈액순환 시 머무는 시 간이 증가하여 전달효과를 높였으며 표적하고 있는 유전자 를 효율적으로 억제 시켰다 (그림 3 d). 또한 높은 혈중 단백 질에의 농도에도 안정성을 보여줬다 [14].
리포좀은 친수성 과 소수성으로 이루어진 리피드의 이중막 구조로 siRNA전 달체로 널리 사용되고 있다. 리포솜은 생체 적합하고 잘 분 해되므로 안전하게 사용할 수 있다. 게다가 인지질이 아민그 룹을 갖고 있으므로 음이온을 띄는 siRNA와 정전기전 결합 을 통해 쉽게 복합체를 형성할 수 있다. 복합체의 형성으로 엔도사이토시스를 원활하게 하여 세포 내 통과를 용이하게 할 수 있다. 또한 엔도좀/라이소좀으로부터 방출이 용이하므로 세포질로 손쉽게 이동할 수 있다. 이와 같이 양이온 리 포좀 과 폴리머를 이용한 siRNA 전달체가 효과적으로 유전 자를 억제하지만 비특이적으로 다른 세포에도 작용하여 부 작용을 초래할 수 있다. 따라서 세포 특이적 전달을 위해 항체, 앱타머, 펩티드, 엽산과 같은 작은 분자들을 전달체에 리간드로 결합하여 표적 세포에만 전달 하는 연구가 진행되 고 있다 [15] (그림 3 e). 또한 exosome을 매개로 한 siRNA 전달 시스템, oligoncolotide 나노 입자 전달체, RNAimicrosponges 등과 같은 새로운 siRNA 전달체들이 많이 연구되고 있고 siRNA 전달에 있어서 충분한 가능성을 시사 해 주고 있다 [16-17].
3. RNAi 치료제 현황
siRNA 전달체의 개발로 전신 투여를 위한 많은 수의 siRNA 약물이 증가하였다. 2004년 처음으로 siRNA를 이용한 RNAi 치료제가 임상시험을 시작한 이 후로, 20종 이상의 siRNA 치 료제가 임상 시험을 진행하고 있다 [18]. 다음 표 1에서는 현 재 외국에서 임상시험 단계별로 진행 되고 있는 siRNA 치료제 들을 정리해 놓았다.
AMD, DME, NAION 과 같은 국소질환 및 바이러스 감염 질환 (RSV, HCV)은 siRNA만 국소적으로 주사하였다.
다른 몇몇의 siRNA 치료제들은 off-target 효과 로 임상실험을 종결시켰다.
Bevasiranib은 임상 3상에서 낮은 치료 효과로 중단되었고, AGN-745 또한 off-target effect 로 임상 2상 단계에서 실험을 중단하였다.
TKM–ApoB는 임 상 1상에서 콜레스테롤의 일시적인 감소현상이 나타나 임상 시험이 종결되었다. 위의 siRNA치료제를 투여했을 때 공통 적으로 TLR (Toll Like Receptor)를 활성화 시켜 면역 부작 용을 일으켰다.
하지만 다른 naked siRNA약물과 달리 I5NP 는 정맥주사 시 임상 2상까지 실시하여 괄목할 만한 치료 효 과를 보여주었다. 많은 선행 연구들은 리포좀을 기반으로 한 siRNA 전달체 플랫폼이 보다 안정적이라고 보고 하고 있다. Nucleic acid lipid particle (SNALP)를 기반으로 한 ALNVSP02 치료제는 임상 1상을 마쳤으며, 일반적으로 간암 환자 의 50%가 항-VEGF 효과를 보여줬다.
또 다른 SNALP 치료제인 ALN-TTR01은 transthyretin (TTR) 단백질이 감소되 었다. 다른 ATTR siRNA 치료제인 ALN-TTR02는 단 한번 의 투여로 TTR 단백질이 94프로까지 억제되는 것을 보여주었 다 [19]. 다른 괄목할 만한 RNAi 치료제로 Atu027은 양이온 리포좀인 AtuPLEX와 결합된 복합체로 임상 1상에서 독성 실 험 결과 33명의 암환자 중 27명에서 부작용이 발생하지 않았 다 [20].
국내에서는 유한양행과 함께 면역 항암제 공동 개발 에 나선 바이오니아가 고효율의 생 분해성 자가조립 나노 입자 인 SAMiRNA 기술을 바탕으로 siRNA 핵심 신약과 신약후보 물질을 개발하고 있다. 이 기술은 RNAi 기술의 단점으로 작용 하던 off-target effect를 줄이고 간 독성 역시 현저하게 낮은 것으로 나타났다. 여러 차례의 동물 실험을 통해 SAMiRNA 나노 입자를 사용한 종양 크기 성장의 억제가 효과적임을 확인 했고 임상 시험을 준비 중이다. 올릭스 역시 RNAi 기반의 핵 산 약물을 개발하고 있는 바이오텍 회사이다. 올릭스는 기존 siRNA의 면역 반응 유발이나 비 표적 유전자 억제와 같은 부 작용을 감소시킨 비대칭 자가전달 RNAi 기술과 전달체 없이 원하는 세포 내로 RNA를 전달하는 자가전달 RNAi 기술의 원 천 특허를 보유하고 있다. 이러한 원천 기술을 바탕으로 비대 흉터 치료제 OLX101 의 비임상 시험을 진행중이며 특발성 폐 섬유화 치료제 OLX-201 개발을 싱가포르와 공동 진행하고 있다. 인코드젠은 siRNA가 체내의 miRNA로 잘못 인식돼 발 생하는 off-target effect를 줄이기 위해 miRNA의 구조 등을 연구, 6번째 피봇(Pivot)의 위치를 변형함으로써 miRNA로의 작용을 차단하는 siRNA-6pi 기술을 고려대 연구진들과 개발 했고 그 결과를 2015년 네이처 커뮤니케이션 학술지에 게재했 다. 고려대와 인코드젠은 산학협력의 형태로 기술을 이용한 다 양한 질환에 대한 적용을 준비 중이다. 4. RNAi 치료제의 전망 및 결론 siRNA 약물은 서열 특이적으로 유전자 발현을 억제 시킬 수 있으므로 질병을 치료할 수 있는 무한한 가능성을 갖고 있다. 새로운 기술과 연구의 발달로 siRNA의 작용을 변화시켜 치료 가능성을 증가시켰으며 off target effect를 감소 시켰다. 따라 서, 현재 많은 siRNA 표적이 임상연구로 계속해서 진행 중이 며 미래 신약 개발의 새로운 패러다임을 보여주고 있다. 하지만 이런 장점에도 불구하고, RNAi 치료제신약으로 실현되기 위해 여러 가지 문제점들을 극복해야 한다. 우선, siRNA 자체의 화 학적 변형이나 구조적 안정화, 전달체 개발을 통해 생체 내 안 정성을 해결해야 한다. 또한 나노 입자의 사이즈, 표면 전하, 형태를 조절하여 전달 효율을 증가시키는 것이 중요한 사안으 로 여겨진다. 이러한 조건들을 충족시킬 때 보다 안정적이면서 긴 시간 동안 순환 하여 특정 조직에 siRNA를 정확하게 전달 할 수 있을 것이다. 앞으로 좋은 전달 시스템을 구축하여 기존 약물치료의 패러다임을 벗어나 21세기를 선도할 수 있는 RNAi 치료제가 되길 기대해본다.
급성골수성백혈병에서 유전자 변이는 진단이나 잔류병소의 검출 뿐만 아니라 치료에 대한 반응이나 재발 유무, 생존율과 같은 예후 를 예측하는 데에 이용될 수 있으며, 이러한 유전자로는 FrenchAmerican-British (FAB) 분류에 따른 아형에서 특징적으로 나 타나는 t(8;21), t(15;17), t(9;11), inv(16) 등[1]과 그 외 fmslike tyrosine kinase 3 (FLT3)[2], N-RAS[2], p53[3], Wilms' 종양 유전자[4] 등 현재까지 약 110여 종이 있는 것으로 알려져 있다. 이 중 FLT3는 stem cell tyrosine kinase-1 (STK-1) 혹 은 fetal liver tyrosine kinase-2 (FLK-2)로도 알려져 있으며, KIT (CD117), FMS, 혈소판유래성장인자(platelet derived growth factor) 수용체와 함께 class Ⅲ tyrosine kinase 수용체에 속 하고, 특징적인 구조적 상동성을 가진다. 즉, 세포외 부위에 5개의 면역글로불린이 고리 형태를 이루는 영역, 세포내 막곁(juxtamembrane) 영역, 2개의 tyrosine kinase 영역(tyrosine kinase domains; TKD)으로 구성되며[5], 조혈모세포에 현저하게 표현되 어 세포의 생존, 증식, 분화, 세포자멸사 등에 관여한다[6-8]. 관 련 유전자는 13번 염색체의 장완(q12)에 위치하며, 24개의 엑손 (exons)으로 구성된다[9].
FLT3 유전자 변이는 급성골수성백혈병에서 가장 흔한 것으로 세포내 두 번째 TKD (D835) 돌연변이와 internal tandem duplication (ITD) 변이의 두 가지 형태가 알려져 있다. D835 돌연변 이는 exon 20에서, ITD 변이는 exon 14와 exon 15 사이에서 나 타나며[10-20], 모두 체세포돌연변이로 서로 독립적으로 발생하고, 같이 나타나는 경우에도 서로 다른 대립형질에서 발생되는 것으로 보고되고 있다[11, 12]. 이러한 FLT3 유전자 변이는 조혈계의 많은 세포들에서 생성되는 배위자(ligand)에 의존하지 않는 수용 체 활성을 유발하여 세포 증식을 초래할 뿐만 아니라 세포자멸사 를 억제하는 데도 관여한다[6-8].
D835 돌연변이는 835번 codon이 정상적으로는‘GAT'로 aspartic acid를 만들게 되나 이 부위가 결손 됨에 따라 aspartic acid 가 결손 되거나 혹은‘TAT' 등으로 돌연변이가 생김에 따라 tyrosine, histidine, valine, glutamate 등으로 치환되며, 836번 codon 의 isoleucine이 결손 되거나 methionine 등으로 치환되는[6, 10- 13] 과오돌연변이로 정상인에서는 발견되지 않는다[10-12]. 이를 분석하는 방법으로는 유전자 부위를 중합효소연쇄반응(polymerase chain reaction, PCR)법으로 증폭 후 제한효소분절길이다형성 분석과 염기서열을 분석하는 방법이 이용되며[10-13, 15], 급성골 수성백혈병에서 이 변이의 의의는 백혈병유발에 관여하거나 질환 의 진행에 어떤 역할을 할 것으로 보고되었으며[10], 백혈구 수나 백혈병 세포의 백분율과 관련되는 보고[13]도 있으나 흔히 생존 율에 영향을 미치는 예후적 의미는 없는 것으로 보고되고 있다 [10-12].
ITD 변이는 막곁 영역에서 발생하는데 exon 14와 exon 15 사 이에서 나타나며[9], 그 크기나 위치는 다양한 것으로 알려져 있 다. 대개 그 크기는 12‐204 bp로 보고되며, 흔히 발견되는 염기 서열은 없으나 589, 591, 597, 599번 tyrosines 중 적어도 하나를 포함하는 것으로 알려져 있고 하나 이상의 ITD 변이가 존재하는 경우도 보고되고 있다[2, 6, 14, 17]. 이 변이를 분석하는 방법으 로는 유전자 부위를 PCR법으로 증폭 후 염기 서열을 분석하는 방 법이 이용된다[12, 13, 15-20]. 이 변이의 처음 발견 시 백혈병유 발과 관련되거나[17] 백혈병의 진행에 중요한 역할을 할 것으로 보고되었다[18]. 그 후 여러 연구에서 백혈구증가증, 백혈병 세포 백분율이 높은 것과 관련되거나[6, 12, 13, 18], 전체 생존율, 무병 생존율, 재발과 관련되는 등의 예후적 의미를 가지는 것으로 보고 되고 있어[2, 12-14, 16, 19, 20] 급성골수성백혈병에서 통상적인 검사로 시행되어야 한다는 주장도 있다[20]. 또한 이 두 가지 변이는 염색체 분석 결과, 정상 핵형인 경우 좀 더 흔히 관찰되므로 이러한 환자 군에서 위험도를 예측할 수 있다 는데 더욱 예후적 의미를 가진다[12, 13, 20]. 그러나, 한국인을 대상으로 한 백혈병에서의 FLT3 유전자 변 이의 예후적 의미에 관한 연구가 없기에 저자들은 급성골수성백혈 병에서 FLT3 유전자의 D835 돌연변이와 ITD 변이 두 가지를 분 석하여 그 빈도를 알아보고 유전자 변이 유무에 따른 예후적 인자 및 생존율, 치료에 대한 반응과의 관련성을 분석하여 그 예후적 의 미를 알아보고자 하였다.
RNA 간섭(RNA interference, RNAi)의 개념을 이용한 치료방법은 매우 촉망받고 있다. 사람 유전체 데이터를 활용하여, mRNA 유전자 서열을 기반으로 한 올리고뉴클레오티드 치료제를 디자인할 수 있다. 표적 서열을 잘 정하면 각각의 모든 유전자들은 RNAi 작용기전을 통해서 억제될 수 있으며, 나아가 전사체, 돌연변이, 스플라이싱 변이체까지 구별하여 표적화할 수 있다. 전통적으로 활성이 있는 물질을 찾기 위해 복잡한 탐색과정이 필요했던 것과 대조적으로, 유전자의 기능에 기반하여 빠르고 쉬운 치료제의 개발이 가능하다.
올리고뉴클레오티드 치료제는 그들의 작용기전에 따라 다음과 같이 여러 가지로 분류된다: 안티센스(Antisense), 압타머(aptamer), 스플라이싱 교정 올리고뉴클레오티드(splice correcting oligonucleotide), siRNA, miRNA 유사체와 길항제, 면역활성조절물질(immunomodulating agent). 세포 내 RNAi 작용 복합체가 매우 효율적으로 작용하므로 siRNA와 miRNA 올리고뉴클레오티드가 치료제로써 가장 각광받고 있다. 일부 대비되는 생물학적 효과에도 불구하고, 치료제 개발의 측면에서 중요한 특징들은 모든 올리고뉴클레오티드 분류에서 공통적이다. 올리고뉴클레오티드는 적어도 전통적인 작은 분자 약물에 비해서 상대적으로 큰 분자이다. 하지만 단클론 항체(monoclonal antibody)와 같은 대부분의 단백질 분자보다는 크기가 작다. 또한, 다중음이온성(polyanionic), 친수성(hydrophilic)의 성질로 인해서 생체 내 분포와 세포 내 흡수에 있어 부정적인 영향이 크다. 화학적 구조와 약학적 제제 또한 약동학적, 약력학적 특성에 본질적인 영향을 미친다.
전사과정에 작용하여 유전자 침묵(gene silencing)을 일으키는 올리고뉴클레오티드는 현재 치료가 불가능한 여러 가지 암, 바이러스감염, 자가면역질환, 심혈관계 질환 등에 새로운 양식의 치료법이 될 수 있다. 치료제로써의 가능성이 알려진 것은 수십 년이 지났지만 현재 단지 2가지의 올리고뉴클레오티드 치료제만이 시장에 나와있다. 또 한가지는 승인되었지만 더 이상 판매되지는 않는다.
최초로 허가를 받은 약은 fomivirsen으로, CMV 바이러스(cytomegalovirus) 감염을 치료하는 안티센스 올리고뉴클레오티드이다. 안내주사(intraocular injection)를 통해 국소적으로 적용된다. 효과적인 HIV 치료요법의 도입으로 인한 낮은 수요로 인해 fomivirsen은 더 이상 사용되지 않는다.
Pegaptanib은 압타머로, 노화에 따른 황반변성(macular degeneration)의 국소 치료에 사용가능하며, VEGF(혈관내피성장인자, vascular endothelial growth factor)의 유해한 효과를 억제한다.
Mipomersen은 안티센스 올리고뉴클레오티드로 2013년에 FDA 승인을 받았으며, 가족성 고콜레스테롤혈증(familiar hypercholesterolemia)의 치료에 사용한다. Mipomersen은 apolipoprotein B100(아포지질단백질 B100)의 발현을 억제하여 혈중 콜레스테롤, LDL-C, apolipoprotein B100의 레벨을 낮춘다. Mipomersen은 최초로 전신적으로 적용된 올리고뉴클레오티드로 대표되기도 한다. Mipomersen은 주로 간세포에서 작용하는데, 그 화학적 변형으로 인해 간 조직 내의 높은 축적이 가능하다. 혈장단백질과 강하게 결합함으로써 신장으로의 배출(신 배설, renal elimination)이 방지된다. Mipomersen이 ALT (liver alanine transaminase), AST (liver aspartate transaminase), 지방간증 (hepatosteatosis)을 증가시키는 것 같은 경향이 나타나 승인절차 도중 논란이 일었다. 결과적으로 mipomersen은 미국에서는, 특별한 제한 하에 심각한 가족성 고콜레스테롤혈증에 대한 치료제로 승인되었다. 유럽에서는, 장기간 사용에 대한 독성에 대한 우려로 EMEA 승인을 받지 못했다.
2세대 안티센스 치료제로써의 Mipomersen은 phosphorothioate와 2’-methoxyethyl 변형을 갖고 있다. Phosphorothioate 변형은 안티센스 계열에서 가장 널리 사용되는 화학적 변형으로, 인산기의 중합하지 않은 산소원자 하나가 황으로 대체된다. 이 작은 화학적 변화는 약리학적 특성에 큰 영향을 미친다. 분해효소에 대한 안정성이 증가되고, 높은 비율로 혈장단백질과 결합한다. 반대로 비특이적 단백질 친화도의 증가로 off-target effect가 나타날 수 있다. Off-target effect란 목표했던 유전자와 다른 표적에 영향을 미쳐 나타나는 부작용을 의미한다. Mipomersen을 포함하여 다른 안티센스 치료제에서도 이러한 Off-target effect는 실제로 상당한 부작용으로 나타남이 확인되었다. 특히 phosphorothioate는 주사부위, 간과 같이 약물의 농도가 높은 곳에서 독성을 유발한다. 안티센스 계열의 약물에서 논란이 되는 여러 문제들이 phosphorothioate 변형에서 유발되는 것으로 지금은 잘 알려져 있지만, 적절한 대체방법이 아직 없다. 뉴클레오티드의 리보오스(ribose)에 변형을 가하는 것(2’-methyl, methoxyethyl, other alkyl chain, locked nucleic acid (LNA))은 분해효소에 대한 안정성을 증가시키는데, 단백질 결합과 세포 내 흡수에 필요한 부분적인 phosphorothioate 변형과 함께 할 때에 효과적일 수 있다. 현재 임상평가 중인 차세대 안티센스 화합물은 비슷한 독성이 나타날 것으로 생각되지만, 높은 안정성 확보로 투여량이 감소하게 되어 독성이 낮게 나타날 것으로 예상된다.
스플라이싱 교정 올리고뉴클레오티드는 유전자 돌연변이 또는 잘못된 유전자 스플라이싱으로 인한 희귀질환에 고무적인 효과를 나타냈다. 희귀질환 Duchenne 근위축병(Duchenne muscular dystrophy)은 비기능성 dystrophin 단백질의 발현으로 유발된다. 유전자의 일부 결실로 인한 프레임시프트(frame-shift)가 나타나고 비기능성 단백질이 발현되는 것인데, 올리고뉴클레오티드로 스플라이싱되는 자리를 막아줌으로써 리딩프레임(reading frame)을 복원할 수 있다. 짧지만 기능성 단백질이 발현된다. 두 가지 올리코뉴클레오티드가 함께 개발되었는데, 한가지는 phosphorodiamidate morpholino oligomer (PMO)인 eteplirsen이다. 나머지 한가지 drisapersen은 임상 2상까지는 거의 부작용이 없는 결과를 보여줬으나, 임상 3상에서 임상적 이점이 크게 나타나지 않는 것으로 나타났다. 추가적인 시험이 진행 중이다.
현재까지 siRNA (short interfering RNA)는 아직 승인 받은 약물이 없다. RNA 간섭 현상이 1998년에 발견되었다는 사실을 고려하면 놀라운 것이 아니다. RNA 간섭의 작용기전을 밝히고, 서열의 디자인, 길이, 화학 변형 및 제제에 대한 연구에 10여년이 걸렸다. siRNA 약물에 대한 임상 평가는 최근 몇 년 사이 시작되었으며, 향후 수년 이내에 추가적인 몇 가지 유망한 약물들이 임상시험에 들어갈 것으로 예상된다.
2. siRNA 치료제 – 간에 존재하는 표적에 대한 효율적인 억제
초기 siRNA 치료제는 눈에 국소적으로 사용하는 데에 개발의 초점을 맞추었다.
***Bevasiranib은 Anti-VEGF siRNA로, 제형화되지 않은 상태로 임상 3상 시험까지 받았다. 하지만 임상반응이 낮게 나타나 임상시험은 곧 종료되었고, 더 신중한 전임상 단계의 필요성이 대두되었다.
siRNA 치료제에서 해결되지 않은 주요 쟁점은, 물질의 안정성, 세포흡수, 엔도좀 탈출(endosome escape), 및 약동학적 특성과 관계된 것들이다. 변형되지 않은 siRNA는 원래 체내에 존재하지 않는 외인성 핵산으로써, 체액 내에 존재하는 효소들로 즉시 분해된다. 리보오스나 인산기의 다양한 화학적 변형을 통해서 체내 안정성을 증가시킬 수 있고, 핵산분해효소로부터도 보호될 수 있다. 다양한 변형으로 체내 안정성을 증가시킬 수 있지만, RNAi 복합체에 의해 조절되는 약동학적 효과를 손상시키지 않는 경우는 소수에 불과하다. 치료용 siRNA에 가장 유용한 변형은 제한된 수의 말단에 phosphorothioate를 도입하는 것과, siRNA 서열 전체에 분산하여 2’-O-methylation 또는 2’-F-nucleotide를 도입하는 것이다. 이러한 변형을 통한 구조적인 최적화는 siRNA의 체내 안정성을 증가시키고, 효소로부터 분해를 방지하므로 siRNA 자체가 약물로 사용될 수 있도록 돕는다. 신장을 통한 신속한 여과 및 배설은 피할 수 없는 과정이다. 안티센스 물질을 통한 많은 연구에서 phosphorothioate backbone 형태의 안티센스는 배설을 회피하고, 세포 흡수를 증가시킬 수 있음이 밝혀진 바 있다. 이와는 대조적으로 siRNA backbone의 너무 많은 변형은 RNAi 복합체를 통해 siRNA의 활성이 나타나는 것을 어렵게 만든다. 결과적으로, 화학적 변형을 통해 siRNA를 적절하게 안정화 시킬 수는 있지만, 리간드 접합 또는 약물전달을 위한 패키징방법 등의 추가적인 접근이 필요하다고 할 수 있다.
세포 흡수와 약동학적 특성의 개선을 위한 방법으로 다음 두 가지 방법이 각광받고 있다: 조직특이적인 흡수 및 축적을 증가시키기 위해 특정 수용체에 특이적인 리간드를 접합하는 방법과, 지질 입자 내에 패키징하는 방법이다(표 1). 지질 입자 내에 패키징하는 방법은, 수년에 걸친 지질 구조 및 조성에 대한 단계적인 발전을 통해 in vitro 및 in vivo 효율이 향상되고 있다.
***현재 가장 진보된 형태의 siRNA는 Alnylam(앨라일람) 사의 patisiran (ALN-TTR02)이며, tranthyretin (ttr)유전자를 표적으로 하는 LNP 패키징된 siRNA이다. Patisirna는 transthyretin에 아밀로이드증(잘못 접힌 단백질에 의한 아밀로이드 침착물이 축적되는 희귀질환)을 치료하기 위해 만들어 졌다. 이 병은 돌연변이의 위치에 따라서 다발성 신경병증 또는 심근병증의 형태로 나타나고, 심근병증의 경우 심부전으로 이어진다. 임상 2상 시험에서 Patisiran은 정상 ttr 유전자와 돌연변이 ttr 유전자의 발현을 모두 효율적으로 감소시켜서 TTR 단백질의 생성을 감소시키는 것으로 나타났다. 단일 투여 후 수주 동안, 또는 다중 투여 후 6개월 동안 TTR 단백질의 발현이 80%까지 감소되었다. 운동 및 감각 신경 기능 및 심장 기능에 대한 임상효과를 평가하기 위한 임상 3상 시험이 2013년에 시작되었다.
***앨라일람에서 TTR-아밀로이드증에 의한 심근병증을 치료하기 위해 피하투여가 가증한 an-ti-TTR-siRNA를 개발 중에 있다. 리간드로 N-acetylgalactosamine (GalNAc)가 siRNA의 센스가닥(sense strand)에 접합되어 있고, asialoglycoprotein 수용체를 통한 수용체 매개 엔도사이토시스가 가능하다. 사람에서 임상 1상 시험에서 10mg/kg siRNA 접합체를 투여하였을 때, 용량의존적 방식으로 TTR 단백질이 최대 94%까지 감소하였다. 3가 GalNAc의 접합(즉 하나의 siRNA 분자에 3개의 탄수화물 분자가 결합)으로 수용체에 대한 높은 친화성이 나타나고, Kd값이 감소되며, 2가 접합체에 비해 마우스 간세포에 대한 세포흡수가 약 10배 증진되었다. 생체 내 분포의 경우 투여된 양의 50% 이상이 간에 분포하는 것으로 나타났다. 생체접합체를 사용하여 siRNA를 목표세포에 전달하고 흡수시키고자 할 때는, 세포 외 조직 및 혈액 순환 중 대사 안정성을 증진시켜야 한다. 따라서, siRNA의 화학적 변형은 LNP-매개 약물전달 방법보다 더 중요하다. 말단 phosphothioate 결합과 함께, 여러 핵산 변형(2’-OMe, 2’-F, 2’-deoxy)을 잘 조합함으로써 siRNA의 반감기를 증가시키고, 마우스에서 표적유전자의 발현저해 효율을 증가시킬 수 있었다. 세포 내 안정성이 높아짐에 따라 더 효율적인 RNA 간섭이 일어났기 때문으로 보인다. Alnylam 사는 GalNAc 접합체를 이용한 여러 가지 siRNA 제제를 개발하였다. 혈우병과 희귀 출혈질환에 대한 ALN-AT3, B형 간염 바이러스에 대한 ALN-HBV, 고콜레스테롤혈증에 대한 ALN-PCSsc 등이 있다.
***Arrowhead Research Corporation 사에서는 탄수화물 GalNAc를 anti-HBV siRNA의 간세포로의 표적화와 세포흡수를 위해서 활용한다. GalNAc를 siRNA에 공유결합 시키는 대신, amellitin-유사 중합체로 이뤄진 캐리어 분자에 결합시킨다. 캐리어는 엔도솜 내의 산성 환경에서 분해된다. 이른바 다이나믹 다중접합체(dynamic polyconjugate, DPC)는 추가적인 PEG-마스킹 리간드(PEG-masking ligand)를 사용한다. PEG-마스킹 리간드는 마찬가지로 산성 pH에서 절단 되는데, 양성자 스폰지효과(proton sponge effect)를 통해서 siRNA의 엔도좀 탈출을 증가시킨다(즉 수소이온의 엔도좀 내 유입과, 뒤 따른 물의 유입을 촉진하여 엔도좀이 팽창하여 파괴됨). 처음에 제시된 다중접합체는 불안정한 이황화공유결합을 통한 siRNA와 중합체의 결합에 의존적이었지만, 최근에는 단순히 siRNA와 캐리어를 함께 사용하는 것으로 진화하였다. 전하가 없는 캐리어와 siRNA는 상호작용이 약하기 때문에, 중합체가 siRNA의 흡수에 영향을 미치는지, 또는 단순히 엔도좀 탈출을 증가시키는 보조제로써 기능을 하는지는 명확하지 않다. 형질전환 마우스 모델에서 혈청 내 바이러스 마커가 1% 미만으로 효과적으로 감소되었다. 사람의 경우 최근 임상 2상 시험이 시작되었다. 2mg/kg 용량의 siRNA 투여 후 바이러스 마커가 약 50%까지 감소되었음이 보고되었고, 고용량에서의 결과는 아직 보고되지 않았다.
최근 microRNA (miRNA)에 지식이 넓어지면서, 치료제로 응용하기 위한 miRNA 유사 올리고뉴클레오티드의 개발이 활발히 진행되고 있다. Phosphorothioate/LNA-변형 단일가닥 올리고뉴클레오티드 miravisen은 HCV 바이러스 복제에 필수적인 miR-122와 HCV RNA의 상호작용을 차단한다. Pre- 및 Pri-miR-122 전구체와의 결합을 통해서 정상적인 miRNA 가공과정을 저해하는 것 또한 약효에 기여하는 것으로 보인다. Miravisen은 현재 HCV 바이러스 감염에 대해 임상 2상 시험이 진행 중이다.
많은 siRNA, miRNA, 안티센스 기반 치료제들이 간 조직에서의 표적 억제효과를 나타냈다. 조직 특이적인 세포흡수와 축적에 있어서 가장 성공적인 방법은 수동적(지질 입자) 또는 능동적(asialoglycoprotein 수용체 리간드)으로 간세포를 목표로 하는 것이었다. 지질 입자는 주로 그 크기 때문에 간으로 직접 유입되며, GalNAc 접합체는 주로 간세포에 발현되는 수용체를 표적으로 한다. 이들 약물들은 siRNA 기술의 효능을 증명하고, 요구되는 투여량, 표적 유전자의 검증, 해당 임상효과에 등에 대한 정보를 수집하는 도구가 될 것이다. 또한 포괄적인 약물 안전성 평가는 모든 종류의 siRNA 치료법에 영향을 미칠 것이다. 치료에 더욱 적극적으로 활용하기 위해서는 간 이외의 조직 및 종양으로 전달을 확장할 수 있는 방법의 개발이 필수적이다.
siRNA 올리고뉴클레오티드의 적용 대상은 모든 유전자 전사체로 확장될 수 있다. 대부분의 전통적인 소분자 약물은 단백질의 특정 결합자리를 표적으로 한다. 때문에 약물이 적용 가능한 표적이 제한된다. 소분자 약물의 질병 관련 표적은 약 1000개 정도로 추정되며, 그 중 30% 미만이 현재 치료를 위한 표적으로 이용되고 있다. 치료용 항체가 등장하면서 세포 밖 표적들까지 약물 표적이 확장되었다. siRNA는 유전자의 번역과정을 방해하기 때문에, 모든 유전자 산물이 약물 표적이 될 수 있다. 일반적으로 소분자 약물보다 개발과 생산 비용이 많이 들고, 약동학적 특성들과 관련하여 해결해야 할 점들이 있지만, 소분자 약물로는 해결할 수 없었던 표적까지 작용할 수 있다는 것이 가치가 크다고 할 수 있다. 하지만, siRNA는 현재 유전자 번역을 억제하는 방향으로만 작용한다. 따라서, 표적 유전자의 과발현과 관련되고, 다른 치료법이 없는 질병을 목표로 약물개발을 진행하는 것이 바람직하다.
분자수준에서의 특이성에도 불구하고, 올리고뉴클레오티드 치료제는 부작용이 선험적으로 배제된다는 의미에서 마술총알(magic-bullet)은 결코 아니다. 서열특이적 또는 서열비특이적 off-target 효과들은 잘 연구되어 있다. 서열특이적인 off-target 효과는 의도한 표적 유전자 외의 유전자와 결합함으로써 나타나는데, 올리고뉴클레오티드 서열을 세심하게 디자인함으로써 막을 수 있다. 서열비특이적 off-target 효과는 단백질과의 결합, 특히 Toll-like 수용체(Toll-like receptor)와 같은 면역활성 수용체와의 결합 등에 의해 유도된다. 부작용은 질환과 관계없는 기관이나 조직에서 표적유전자를 너무 강하게 침묵시킴으로써 나타날 수도 있다. 예를 들면, 암세포를 표적으로 공격할 때, 종양에서의 특정 유전자가 과발현되는 것을 표적으로 침묵시키는데, 정상세포에서 기본적으로 발현되는 표적 유전자도 억제될 수 있음을 고려해야 한다. 결과적으로, 다른 치료제들과 마찬가지로 siRNA의 표적지향적인 전달은 효능과 낮은 부작용을 위해서 siRNA의 서열 특이성 못지않게 중요하다고 할 수 있다. 물론 siRNA의 표적지향적인 전달특성은 다른 약물들의 전달특성과 많은 측면을 공유하지만, 올리고뉴클레오티드 전달에 특이적인 측면들도 있다. 다중음이온성 중합체인 핵산은 효율적으로 패키징되고 분해로부터 보호될 필요가 있다. 성공적인 세포흡수 이후에는 세포질로 잘 방출되어야 한다. 대부분의 저분자 화합물 약물들의 경우 세포질로의 수송이 약효에 결정적이지 않은 것과 대조적이다.
2.siRNA therapeutics — efficient reduction of liver targets
Just as prior efforts with therapeutic antisense andaptameroligonucleotides, initialsiRNAdevelopments focused on local administration to the eye.
***An anti-VEGF siRNA,bevasiranib, was evaluated in a phase III trial after application of the unformulated drug[29]. However, poor clinical responses soon led to termination of this trial, emphasizing the need for thorough and careful preclinical development instead of rushing a poorly designed agent to clinical trials. The main issues yet to be resolved are substrate stability, cellular uptake and endosomal escape, andpharmacokinetics[30],[31]. Being an exogenousnucleic acid, unmodified siRNA is prone to nearly instant enzymatic degradation in body fluids.
Variations of thechemical structureat theriboseor the phosphate backbone can significantly increase the stability, and proper modification patterns result in negligible cleavage bynucleases. Although many modifications are useful for increasing stability, only few of those are also apt for keeping intact the pharmocodynamic effect modulated by the RNAi machinery. For therapeutic applications, the most useful siRNA modifications are a limited number of terminalphosphorothioatelinkages, and several 2′-O-methyl and/or 2′-F-nucleotides scattered throughout the sequence[32],[33]. These structural optimizations allow the use of unpackaged siRNA by increasing enzymatic stability and preventing nuclease cleavage. Rapid renal filtration and elimination are not avoided[33],[34],[35],[36]. Ample experience with antisense compounds has proven that modification with a phosphorothioate backbone evades elimination and increases cellular uptake[17]. In contrast to the antisense technology, a high degree of backbone modifications renders siRNA oligonucleotides unable to activate the RNAi machinery. Consequently, chemical modifications aptly stabilize siRNA, but need an additional targeting approach, either through ligand conjugation or by packaging in a delivery system.
For improvement of the pharmacokinetic properties and cellular uptake, two distinct approaches seem to be most promising: the direct attachment of receptor-targeted ligands to the oligonucleotide for increasing tissue-specific accumulation and uptake[37], and the packaging in lipid particles[38](Table 1). For the latter approach, stepwise evolution of lipid structures and particle composition has resulted in improvedin vitroandin vivoefficiency over the years[39]. The currently most advanced siRNA in the clinic is Alnylam'spatisiran(ALN-TTR02), an LNP-packaged siRNA targeted at thetransthyretin(ttr) gene[3]. It is developed for treatment of transthyretin mediatedamyloidosis, an orphan disease in which mutated proteins are misfolded and build up amyloid deposits. Depending on the mutation's locus, the disease results inpolyneuropathyand/orcardiomyopathy, the latter form culminates in progressive heart failure. Patisiran efficiently knocked down both wild-type and mutatedttrgenes and thus reduces TTR protein production in phase II trials. A sustained effect of an 80% TTR reduction was achieved for several weeks after a single dose and six months after multiple dosing. The multi-center phase III APOLLO trial for evaluation of clinical effects onneurological symptoms, motor and sensory nerve functions and heart function has begun in late 2013.
Table 1.Selection of ongoing clinical trials ofsiRNAagents. Respective delivery systems and chemical modifications as well as administration routes are listed for illustration of early development (focusing on local administration of unformulated siRNA), and ongoing trials (systemic applications, primarily for liver-associated diseases).
The same company is developing an anti-TTR-siRNA sequence as a bioconjugate for subcutaneous application for treating TTR-amyloid-induced cardiomyopathies. As a targeting ligand,N-acetylgalactosamine(GalNAc) is attached to the sense strand of siRNA using a trivalent linker to achievereceptor-mediated endocytosisvia theasialoglycoprotein receptor[40],[41],[42]. In a phase I test in humans, TTR was reduced in a dose-dependent manner with a maximum of up to 94% reduction after application of 10 mg/kg siRNA conjugate. The trivalent GalNAc linkage,i.e.attachment of three molecules of the targeting carbohydrate to one siRNA molecule, ensures high affinity to the receptor, decreases the Kdand thus enhances cellular uptake in mouse hepatocytes around ten-fold compared to adivalentconjugate[42].Biodistributiondatain vivosuggest that a high proportion (> 50%) of the applied dose is delivered to the liver. When usingbioconjugationto afford siRNA targeting and uptake, the metabolic stability needs to be increased because of the lack of shielding in extracellular tissues and blood circulation. Thus, chemical modification of the siRNA is more important than for LNP-mediated delivery. A well-attuned combination ofnucleotidemodifications (2′-O-Me, 2′-F, 2′-desoxy) together with several terminal phosphorothioate linkages not only increases the half-life of the agents, but also enhances the target knockdown efficiency in mice[42]. This was attributed to the higher intracellular (cytosolic) stability resulting in more efficient RNA interference. The encouraging data prompted Alnylam to develop several other siRNA agents as GalNAc conjugates including ALN-AT3 forhemophiliaand rare bleeding disorders, ALN-HBV forhepatitis B virusinfections, and ALN-PCSsc forhypercholesterolemia.
The carbohydrate GalNAc is also utilized as a targeting agent by Arrowhead Research Corporation for achieving targeting to and uptake into hepatocytes of their anti-HBV siRNA[4]. Instead of covalently attaching GalNAc to the siRNA agent, the ligand is tethered to a carrier molecule, a mellitin-like polymer that is degraded in the acidic environment of endosomes. This so called dynamic polyconjugate carries additional PEG-masking ligands that are likewise cleaved at acidic pH-values, and release the previously masked cationic charge of the polymer. This sophisticated design is supposed to increase endosomal escape of the siRNA through the proton sponge effect,i.e.induction of influx of hydrogen ions followed by water and swelling of the endosomes, finally causing their disruption[43]. While initially presented data on polyconjugates relied on a covalent, but labile attachment of the siRNA cargo to the polymer via adisulfidelinkage, a more recent evolution of the systems uses simple co-application of siRNA and the carrier[44]. Because little interaction of the uncharged carrier with siRNA can be expected, it is not fully clear to what extent the polymer influences direct siRNA uptake or if it is simply an adjuvant for increasing endosomal escape. In atransgenic mousemodel, efficient reduction to less than 1% of serum viral markers was detected[4]. Evaluation in humans in a phase II trial has lately been initiated. Early data that have been disseminated indicated reduction of viral markers to around 50% of the baseline after administration of siRNA at a 2 mg/kg dose. The results of higher doses are not yet published.
The recently expanding knowledge aboutmicroRNAs(miRNA) has quickly prompted the development of miRNA-mimicking oligonucleotides for therapeutic applications[45]. A respective antagonizing phosphorothioate/LNA-modified single stranded oligonucleotide,miravirsen, blocks the interaction of humanmiR-122with HCV RNA, an essential step in viral replication[46]. An additional binding to precursor pre- and pri-miR-122 structures and consequent inhibition of nuclease-mediatedmiRNAprocessing seem to contribute to the pharmacological effect[47]. Miravirsen is currently undergoing phase II evaluation against HCV infections.
It is no surprise that all these clinically evaluated siRNA and miRNA drugs, as well as many antisense agents, are tested for the antagonization of liver targets[48]. The most viable approaches for tissue enrichment and cellular uptake make use of passive (lipid particles) or active (asialoglycoprotein receptor ligand) hepatocyte targeting. Lipid particles are directed to the liver mainly because of their size, and GalNAc conjugates target the respective receptor mainly expressed on hepatocytes. These drugs will be instrumental to prove the principal efficacy of the siRNA technology and gather information about required dosing, target validation, achieved level of gene expression reduction and corresponding clinical effects. Furthermore, comprehensivedrug safety assessmentswill have implications for the whole class of siRNA therapeutics[49],[50]. For making fulltherapeutic useof the potency of the technology, it is essential to develop modalities for expansion of the target space to other organs, tissues, and tumors.
Because of their mode of action, siRNA oligonucleotide agents instantly broaden the druggable target space to all gene transcripts. Nearly all traditional, small molecule drugs rely on distinct binding pockets of proteins. Consequently, the number of potentially druggable targets is limited. The number of disease-relevant targets for small molecules is estimated at around 1000, with less than 30% of that number currently being exploited for therapy[51]. The advent of therapeutic antibodies has extended the druggable target space, but only to extracellular molecules. Because of interfering with translation,siRNAscan target any desired gene product. Because of the challenges associated with the pharmacokinetics and generally higher costs for development and manufacturing compared to small molecules, the main potential virtue is the antagonization of targets that are not available for small molecules. However, siRNAs currently only work in one direction, that is inhibition of gene translation. As a consequence, development programs preferably focus on diseases unavailable for other treatment options, and on targets with validated overexpression and disease-relevance.
Despite their intrinsic specificity on the molecular level, gene-silencing oligonucleotides are by no means magic bullet in the sense that side effects are excludeda priori. Both sequence-specific and sequence-unspecific off-target effects are well-documented[19],[52],[53],[54]. While the first, caused by hybridization to nucleic acid targets other than the intended, can be avoided by careful oligonucleotide design, the latter is induced by binding to proteins, particularly immune-activating receptors such astoll-like receptors[53]. Side effects can also arise from excessive silencing of the target, especially in non-disease relevant organs and tissues. For example, when attacking a cancer target, it is assumed that the exaggerated expression in tumors is responsible for a higher impact of therapeutic silencing than that in healthy cells. However, since a certain basal expression exists in somatic tissue, the interference with gene expression in unaffected tissue is a cause for concern. Consequently, targeted delivery of siRNA is an important aspect for adding a second layer of specificity, and like for any other therapeutic agent, promises better efficacy with lower adverse effects. Of course, many issues and formulations of targeted siRNA delivery are shared with delivery of other drugs, but there are some specific aspects for oligonucleotide transport: The polyanionic nucleic acid cargo needs to be efficiently packaged and shielded from degradation. After successful cellular uptake, the oligonucleotides must be released into the cytosol in order to exert their therapeutic effect. This is in contrast to most low-molecular weight compounds, for which successful cytosolic transport is often not decisive.
2.1 생체 장벽 및 생체 내 분포(Barriers and biodistribution)
siRNA 치료제가 그 작용 장소인 세포질에 다다르기 위해서는 많은 생체 장벽들을 극복해야 한다. 전신투여(정맥투여 등) 후 siRNA 치료제는 혈액순환을 거쳐 각 장기에 분포하게 되어 신장으로의 배설을 피하게 된다. 각 장기에 도달한 siRNA 화합물은 혈관내피세포 장벽을 뚫고 간질로 이동해야 한다. 간질을 이동한 siRNA는 표적세포에 결합하고 엔도사이토시스(endocytosis)를 통해 흡수된다. 흡수된 siRNA 화합물은 엔도좀을 뚫고 나와야 RNAi 복합체와 만날 수 있다. 이러한 모든 과정은 여러 가지 투여방법에 따라 큰 영향을 받고, 적절한 해결책이 필요하다.
정맥주사 외에도 피하주사, 피내주사, 폐 또는 코 점막을 이용한 투여방법은 혈류를 통하게 된다. 하지만 이러한 방법들은 추가적으로 극복해야 할 장벽을 수반하므로 현재의 임상적인 시도들은 피하주사 또는 정맥 내 주사(완전한 흡수가 예상되는)에 집중되어 있다. 우선 피하주사 또는 정맥 내 주사를 통한 효율적인 전달 시스템이 개발되어야 하며, 그 후에 다양한 다른 경로로 확장 적용될 수 있을 것이다.
피부는 넓은 범위에 쉽게 접근할 수 있다는 점에서 매력적인 투여 경로이다. 피부 장벽은 투과성이 없는 각질세포층과, 연속적인 각질층으로 이뤄져 있다. 크기가 큰 친수성 물질을 피부장벽을 뚫고 투여하기 위한 가장 확실한 방법은 미세바늘을 사용한 물리적 방법이다. 치료용 siRNA는 스테인리스 바늘에 코팅하거나 미세바늘장치를 통해 주입할 수 있다. 표피에서의 흡수 및 유전자 침묵효과는 잘 나타났으나, 진피 모세혈관층으로의 흡수는 거의 나타나지 않았다. 피부질환 부위에 바늘을 사용하는 것은 통증을 수반하므로 임상적 사용에 한계가 있었다.
이와 유사하게 siRNA를 폐에 적용하는 방법 또한 국부적인 영향만을 나타낸다. 전임상단계에서 성공적이었던 많은 siRNA들이 실제 임상에서 적용될 수는 없었다. 비강 및 기관 내 흡입을 통한 전달 방법으로, 지질(중성 및 양이온성), 고분자(polyethyleneimine (PEI), poly (lac-tic-co-glycolic acid) (PLGA) 등), 무기물질 캐리어(탄산칼슘, 다공성 실리카 입자) 등이 시도되었다.
혈액 등의 체액 내의 핵산분자는 말단 뉴클레오티드의 인산결합을 절단하는 exonuclease에 의해서 빠르게 분해된다. 핵산을 내부 인산결합부터 절단하는 endonuclease에 의한 분해는 적다. 따라서, 외인성 올리고뉴클레오티드는 말단부위에서부터 단계적으로 분해되어 짧아진 형태의 올리고뉴클레오티드를 형성하게 된다. 본래의 올리고뉴클레오티드와 짧아진 올리고뉴클레오티드 모두 신장 여과를 통해 제거된다. 거대분자의 여과 한계는 약 2~4nm로, 20~40kDa 정도의 핵산에 해단된다. PEG(polyethylene glycol)와 같은 리간드를 부착하거나, 혈장단백질과 결합함으로써 입자의 크기를 크게 만듦으로써 siRNA의 신장 여과를 막을 수 있다.
한편, 크기가 큰 입자는 세망내피계(reticuloendothelial system, RES)에 의해서 차단된다. 일반적으로 200nm가 넘는 입자는 간과 비장에서 주로 발견되는 대식세포에 의해 빠르게 제거된다. 그러나 PEG와 같은 양친매성 물질을 달아줌으로써 RES를 회피할 수 있다.
혈관내피세포는 기관으로의 약물분포를 결정한다. 거대분자와 나노입자는 주로 대류(convection)에 의해 혈관 밖으로 유출되며, 모세혈관을 통과하는 유량이 증가할수록 증가된다. 혈관내피세포 장벽을 가로지르는 정도는 모세혈관의 구멍 크기에 좌우되며, 이는 사용할 수 있는 입자크기의 상한선으로 작용한다. 기관이나 조직에 따라서 혈관내피세포 사이의 접합은 매우 치밀하기도 하고(혈액뇌장벽), 헐겁기도 하다(소화기관의 점막). 간의 경우, 다른 림프조직과 조혈기관과 마찬가지로, 불연속적인 굴모세혈관(sinusoidal capillary)으로 이루어져 있다. 이들은 연속모세혈관(continuous capillary)이나 유창모세혈관(fenestrated capillary)과 달리 구멍을 덮고 있는 막이 없기 때문에, 매우 우수한 투과성을 보이며 거대분자의 교환이 가능하다. 언급한 것처럼 siRNA 전달 시스템의 경우 간으로 잘 전달된다는 특성이 있다. 이것은 지질기반의 입자, 양이온성 또는 중성 나노입자, 지질친화성 리간드 접합체와 관련이 있다. 간 혈관계의 불연속성(나노입자, 단백질에 결합한 올리고뉴클레오티드) 또는 간에서의 지방대사(리포좀, 지질 또는 콜레스테롤 접합체 사용) 때문에 간으로의 축적이 나타난다. 서로 다른 종류의 간세포를 구별할 수 있는 특정 수용체를 표적으로 하는 리간드를 사용함으로써, Kupffer 세포(간에 상주하는 대식세포) 내에만 축적되는 것을 방지할 수 있다. 3가 GalNAc 접합체는 간세포로의 흡수를 증가시키고, 결과적으로 간세포 내에서의 유전자 침묵을 일으킨다. Asialoglycoprotein 수용체는 효율적인 엔도사이토시스를 일으킨다.
몇몇 질병은 조직에서의 모세혈관 투과성을 변화시키기도 하는데, 특히 종양의 경우, 모세혈관의 구멍을 증가시켜 큰 분자와 입자가 통과할 수 있다. 이런 기이한 특징은 EPR 효과(enhanced permeability and retention effect)라고 알려져 있다. EPR효과는 종양에 대한 나노입자의 수동적인 타겟팅에 매력적인 방법이다. 실제로 EPR 효과는 마우스 모델에서 종양세포에 나노입자가 축적되는 것을 설명하기 위해 적용되어왔다. 한편, 수동적인 타겟팅은 종양에서 최대 20%의 축적을 보이며, 혈관 누출 정도는 종양의 유형 등에 따라 다르게 나타난다. 혈관근처의 종양 성장, 종양 내 압력, 및 신생혈관형성 속도는 모두 EPR 효과의 불균등함에 기여하는데, 이들은 종양 모델마다, 개개인의 환자마다, 또는 하나의 종양 내에서도 다르게 나타난다. 동물에 이식한 종양 모델은 사람에서보다 훨씬 빠른 성장을 나타내기 때문에, 혈관의 구멍은 일반적인 전임상시험 모델보다 더 크고, 때문에 EPR 효과가 과장되어 해석되는 경우가 많다. 실제 치료환경에서 나노입자 종양세포 타겟팅에 대한 명확학 임상적 증거는 아직 밝혀지지 않았다.
혈액뇌장벽(Blood-brain barrier, BBB)은 혈관내피세포 사이의 매우 치밀한 접합에서 기인하며, 뇌 조직으로의 자유로운 물질 확산을 방지한다. 치료목적으로는 지질 친화성이 높은 400Da 이하의 분자량 갖는 물질 또는 능동적 수송 시스템이 존재하는 물질만이 뇌에 접근할 수 있다. 생체거대분자 및 나노입자 전달 시스템은 혈액뇌장벽을 통과할 수 없어 치료에 적용하기 어려웠다. 핵산분자를 수송할 수 있는 특정 능동수송 시스템이 없으므로, BBB 트랜스사이토시스를 활용한 전략의 개발이 필요하다. BBB 특이적인 세포 외 마커들이 존재하며, 다른 조직의 혈관내피세포에서도 특이적인 수용체가 발견되기도 한다.
간에만 축적되는 것을 피하고, 다른 기관에서 siRNA를 통한 표적 유전자 침묵을 달성하기 위해서는 수용체 특이적인 리간드의 사용이 필수적으로 보인다.
2.2 세포 내 흡수 및 세포 내 움직임(Cellular uptake and intracellular trafficking)
혈관내피세포를 뚫고 나와 간질을 지나면, 세포막이 기다리고 있다. 어쩌면 가장 어려운 장벽일지도 모르는 세포막을 통과하는 것에 대한 많은 연구가 이뤄졌다. siRNA 자체이건 접합체가 결합된 형태이건 siRNA의 주된 흡수 방법은 엔도사이토시스로 생각된다. 엔도사이토시스가 일어나는 경로는 여러 가지가 있다: clathrin-매개 경로, caveola 의존적인 흡수, clathrin이나 caveola와 관계없는 경로, 피노사이토시스(pinocytosis, 음세포작용). 대식세포, 과립구, Kupffer 세포 등에서는 포식작용이 일어난다. 이들 각 기전들 사이에는 중요한 차이들이 존재하기는 하지만, siRNA 치료제 흡수의 최후는 엔도좀 소포를 이루는 것이다.
siRNA이 기능적으로 활성을 나타내기 위해서는 엔도사이토시스 과정보다 엔도좀을 탈출하는 것이 더 중요하다. 일반적으로 소수의 siRNA 분자만이 엔도좀에서 세포질 내로 옮겨지고, 나머지는 라이소좀(lysosome)과의 융합으로 분해된다. 엔도좀의 세포 내 움직임에 대해서는 완전하지는 않지만 어느 정도 연구가 되어있다. 엔도좀 시스템은 대부분이 세포막으로 다시 회수되는 점이 특징이다. 초기 엔도좀 속의 화합물은 특별한 경로를 통해서 세포 내로 흡수된다. 초기 엔도좀은 세포막으로 다시 회수하거나, 엔도좀을 성숙시켜 후기 엔도좀이 되거나, 라이소좀과 융합하거나, 목표구획으로 엔도좀을 전달시키는 등 엔도좀에 결합된 세포질 내 단백질에 의해 운명이 결정될 수 있다. Rab family, 특히 Rab5는 이 과정의 중요한 조절인자 중 하나이다. 모터 단백질은 actin이나 tubulin 미세소관을 통한 세포 내 표적 소기관으로의 이동을 담당한다. 엔도좀의 세포 내 움직임에 대한 많은 연구에도 불구하고, 엔도좀으로부터 화합물을 탈출시키기 위한 합리적인 수단이 부족하다. 리포좀, 고분자, 수용체 특이적인 리간드를 통해 흡수된 siRNA가 실제로 세포질 내에 도달하는 양이 어느 정도인지 명확하지 않다. 물리적인 기전을 통해 엔도좀 탈출을 증가시키려는 시도는 있었다. 이른바 양성자 스폰지 효과(proton sponge effect)는 염기성 거대분자(펩타이드, 고분자 등)의 산성화를 통해 라이소좀 내에 수소이온이 유입되는 것을 말한다. 양성자의 유입으로 물의 유입과 그에 따른 소포의 팽창이 일어나고 결국 터져서 세포질 내로 화합물이 방출될 수 있다. siRNA의 세포질 내 전달을 증가시키기 위한 다른 전략으로 라이소좀과 엔도좀의 막융합을 이용하는 방법이 있다(산성 pH에서 단백질들의 구조가 변하고 서로 융합하여 투과성이 증가한다). 하지만 이러한 방법은 실제 치료적 조건에서 siRNA 효과의 증가를 나타내지 않았고, 안전 조건도 만족시켜야 한다. 엔도좀의 세포 내 움직임에 대한 지속적인 연구는 올리고뉴클레오티드의 엔도좀 탈출을 개선하는데 도움이 될 것이다.
성공적인 siRNA의 세포 내 전달을 위해서 입자크기, 전하, 안정성, 표면의 접합체, 수용체 특이 리간드의 존재 등 다양한 변수가 있음을 알 수 있었다.
2.3 BBB 트랜스사이토시스(BBB transcytosis)를 위한 표적 및 리간드
종양, 뇌, 혈관내피와 관련된 질환에 대한 siRNA 치료제 전달은 아직 개발이 필요하다. 선택적인 약물전달을 위해서는 장기, 조직, 종양에서만 특이적으로 발현되거나 특별히 많이 발현되는 표면 수용체가 필요하다. 혈관내피 조직간의 유사성과, 종양에서의 발현 패턴의 다양성 등 때문에 이런 수용체를 찾는 것은 쉬운 일이 아니다(표 2).
표 2. 뇌, 백혈구 및 종양조직에 대한 표적화된 siRNA 전달 시스템.
- 수용체 특이적 리간드, 각각의 표적, siRNA 제형(나노입자 또는 생체접합체)을 함께 짝지었다.
BBB를 통과하는 siRNA 전달을 위해서는 트랜스사이토시스를 유발하는 transferrin 수용체가 가장 빈번한 표적이 된다. 인슐린 수용체와 LDL 수용체 또한 BBB에 대한 siRNA 전달에 사용된다. Transferrin 수용체를 표적으로 한 전달방법은 약물의 성공적인 뇌 내 흡수를 보여줬다. 하지만 뇌의 미세혈관 뇌에서 포획되는 케이스도 보고되었다. 최근에는 transferrin 수용체와 리간드의 친화도가 결정적으로 중요함이 보고되었다. 친화성이 높은 리간드는 낮은 리간드에 비해 적은 뇌 축적을 나타냈다. In vivo 이미징을 통해서 친화성이 높은 리간드는 세포 내에서 라이소좀으로 운반되고 분해됨이 밝혀졌다. 때문에 transferrin 수용체 자체도 감소하게 되고, 2차적인 약물의 효과도 감소시킨다.
많은 핵산 전달 시스템에서 뇌 내 축적이 크게 증가하였다. 혈액뇌장벽을 표적하기 위해서 다양한 리간드들(holo-transferrin, transferrin 항체, 변형된 디프테리아 독소, angiopep-2, apoE유사 펩타이드)이 리포좀에 접목되었다. Transferrin 항체를 접목한 리포좀만이 마우스 in vivo 모델에서 뇌 축적을 크게 증가시켰으나, 뇌로 흡수되는 양은 투여량 대비 0.1% 미만이다. 투여된 양의 대부분은 간으로 분포한다. 유전자 전달을 위해서 리포좀은 transferrin 수용체와 인슐린 수용체에 대한 단클론항체 및 PEG 사슬로 수식된 리포좀이 여러 연구에서 사용되었다. 이종이식 신경교종(glioma) 마우스 모델에서 랫(rat) transferrin 수용체 항체와 사람 인슐린 수용체 항체를 도입한 리포좀을 이용했을 때, RNA 간섭을 위한 플라스미드가 종양으로 활발히 운반되었다. 목표했던 EGF 수용체 유전자의 효율적인 침묵이 나타났고 생존기간도 증가되었다. 이러한 접근 방법이 더 많은 축적을 필요로 하는 올리고뉴클레오티드 전달에도 성공할지는 확실하지 않다.
최근에는 siRNA의 축합과 선택적인 뇌 흡수를 위해서 myristoylated transportan과 transferrin 표적 서열이 융합된 펩타이드가 사용되기도 하였다.
BBB를 통과하는 siRNA 전달을 위해 광견병바이러스의 당단백질(rabies virus glycoprotein, RVG) 또한 사용되고 있다. 29개 아미노산의 펩타이드는 광견병바이러스가 숙주세포에 부착, 흡수될 때 필요한 성분이다. RVG와 상호작용하는 수용체와 정확한 전달 기전은 아직 연구되지 않았다. Nicotinic acetyl choline 수용체, 신경세포 접착분자(neuronal cell adhesion molecule, NCAM), 신경 성장인자 수용체(p75NTR) 등이 가능성이 있어 보인다. 이들은 광견병바이러스의 흡수와 감염에 홀로 필수적이지는 않지만, 각각의 돌연변이에서는 바이러스 감염에 대한 감수성을 나타냈다. 연구결과들은 하나 이상의 수용체가 RVG의 흡수에 관여하고 있음을 나타낸다. 밝혀진 수용체는 뉴런세포에 존재하지만 BBB 혈관내피세포에는 존재하지 않기 때문에, RVG 매게 방법이 어떻게 뇌로 운반을 일으켰는지는 명확하지 않다. 광견병 바이러스는 역행성 축삭 수송에 의해 이동됨을 고려할 때, 말초신경세포에 흡수되어 BBB를 넘어서 이동되는 것인지도 모른다.
RVG의 중추신경계 지향적인 특성을 효과적으로 이용하기 위해, Arg 9개와 융합하여 핵산화합물 복합체를 형성하도록 하였다. 이 시스템은 신경세포 특이적이었으며, siRNA 화합물을 뇌에 성공적으로 전달하고 유전자 침묵을 일으켰다. PEI, 리포좀 및 polyamidoamine (PAMAM) 덴드리머를 이용한 비슷한 시스템도 개발됐다. 높은 전하를 띠는 구형 PAMAM 덴드리머에 RVG가 접합되었을 때, 뇌 축적이 크게 증가하였다. 꼬리정맥을 통한 투여 후, RVG가 접합된 리포좀은 높은 뇌 축적을 나타냈지만, 리간드가 접합되지 않은 리포좀은 BBB를 통과하지 못했다. 이황화결합을 통해 RVG를 부착한 PEI 캐리어를 이용했을 때는, in vitro에서 Neuro2a 세포 흡수가 증가되었고, in vivo에서도 뇌 축적이 크게 증가하였다. 큰 크기 때문인지, 간에서의 대식작용을 통한 흡수도 유의미하게 나타났다. 뉴런세포에서의 흡수는 bungarotoxin (nicotinin acetylcholine 수용체에 결합함)에 의해 경쟁적으로 억제됨을 보였다. 흡수는 GABA에 의해서도 억제되었는데, GABAB 수용체가 관여하고 있을 수도 있음을 시사한다.
RVG 펩타이드는 엑소좀(exosome)을 이용한 시스템에도 활용되었다. 엑소좀은 생체 내에서 만들어 지는 소포체의 한 종류로 세포간 small RNA의 수평이동에 관여한다. 세포 내부에서 엔도좀과 비슷하게 형성되어 세포 외부로 엑소좀으로 배출될 수 있다. 다른 소포들과는 다르게 엑소좀은 그 직경이 100nm정도로 작다. RVG 펩타이드를 엑소좀 시스템에 도입하여 정맥투여 하였을 때, 마우스 뇌에서의 siRNA-매개 GAPDH, BACE1 유전자 침묵을 일으킬 수 있었다. 종합적인 생체분포데이터는 없지만, 엑소좀 시스템은 간 축적률도 낮은 것으로 보인다. 앞서 언급한 엑소좀-RVG 시스템에서 간에서는 GAPDH의 발현저해가 나타나지 않았다. 입자의 크기가 작은 특성 때문에 실제로 간에 축적되지 않는 것인지, 또는 단지 siRNA가 간에서 잘 방출되지 않은 것인지는 확실하지 않다. 비강 내 투여를 사용한 다른 연구에서는, 엑소좀(30-100nm)와 마이크로입자(0.5-1um) 모두 간 축적은 나타나지 않았다. 마이크로입자는 폐에 침착되었고 혈류까지 도달하지 못했다. EGFR-엑소좀을 꼬리정맥을 통해 투여하였을 경우는 상당한 간 축적을 나타냈다.
2.4 백혈구와 혈관내피세포에 대한 표적 및 리간드
Integrin은 세포접착분자(cell adhesion molecule)에서 가장 큰 계열로, α와 β 소단위체로 이뤄진 이종이량체(heterodimer)단백질이며, 다양한 생리학적 상황에서 세포-기질, 세포-세포간 상호작용을 중재하는 역할을 한다. 이들은 세포표면에 광범위하게 분포하고 있고, 효과적인 세포내함입(internalization)을 일으키므로 수용체 매개 약물전달의 타겟으로 관심이 높다. 24개의 integrin 계열 중, β2, β7 integrin은 백혈구에서만 발현된다. 포유동물의 α8 integrin은 혈관과, 내장평활근, 신장 사구체 및 폐포에서 특이적으로 높게 발현된다. 랫 폐에서 anti-α8 integrin 항체로 면역염색되는 세포는 폐포 간질에 있는 근섬유모세포(myofibroblast)이다. 폐포 근섬유모세포는 폐포표피와 기저막에 인접하여 모세혈관을 따라 존재한다. 또 다른 integrin 단백질들이 종양혈관계 등에서 발견되기도 한다.
조직에 분포하는 integrin을 살펴보면 integrin의 기능을 추측할 수 있다. 다양한 세포유형에서 광범위하게 발현되는 integrin도 있지만(α2β1, α3β1 등), 제한된 분포를 갖는 integrin도 있다. αIIb 소단위체는 오직 혈소판에서만 발현되며 혈소판 응집의 매개체가 되는 것으로 여겨진다. 종양 혈관내피세포에서는 αVβ3 소단위체의 발현이 증가되어있고, β2과 β7 소단위체는 거의 백혈구에서만 발견된다. 대부분의 integrin의 발현은 발생학적으로 조절된다. 예를 들어 α4β1 integrin은 배아 발달시기 동안 광범위하게 분포하지만, 성인의 경우 백혈구와 내피세포에서 발견된다.
백혈구를 표적으로 하기 위해서 integrin 항체에 기반한 전달 시스템이 개발되었다. LFA-1 integrin(백혈구 특이적인 β2 소단위체를 가짐) 항체를 protamine에 융합하여 siRNA 복합체를 형성하였다. 이 시스템은 LFA-1 수용체를 발현하는 외인성 K562 세포에 특이적으로 siRNA 복합체를 전달하는데 성공하였다. 비슷한 방법으로 β7 integrin 소단위체에 대한 항체를 hyaluronan을 통해 리포좀 표면에 도입하여 백혈구 특이적인 유전자 침묵을 일으켰다. 표지된 지질입자를 사용하여 생체 내 분포를 조사했는데, 염증이 있는 장에서 3.5배정도 분포가 증가하였다. 입자크기가 크므로 간과 비장에서의 현저한 축적도 역시 나타났다.
종양 내피세포에서 증가된 αVβ3 integrin의 발현을 이용하여, cyclic RGD를 도입한 양이온성 지질을 이용한 siRNA 전달이 가능하였다. In vitro에서 αVβ3 integrin은 성공적인 흡수 및 유전자 침묵에 필수적이었고, 실제 In vivo에서 종양내피세포 특이적인 유전자 침묵을 일으킬 수 있었다. 펩타이드 리간드의 변형에 따라 생체 내 분포 패턴이 달랐고, 간에 축적되는 양이 감소하는 만큼 비장이나 폐, 신장에서의 축적이 증가되었다. 하지만 투여량의 50% 이상이 여전히 간에 남아있었다. PAMAM 덴드리머에 RGD 펩타이드가 도입되기도 하였는데, 세포막과의 전하를 통한 결합능력이 강한 덴드리머의 본질적인 성질 때문에, 리간드로 인한 추가적인 세포 흡수의 증가는 나타나지 않았다. 그러나 종양에 대한 더 깊은 침투능력을 나타냈는데, αVβ3과 RGD 펩타이드의 결합으로 세포외 기질과의 상호작용이 저해되었기 때문으로 생각된다.
αVβ3 integrin을 발현하는 세포를 표적으로 하기 위해 siRNA에 Cyclic RGD 펩타이드가 결합되었다. RGD의 이온가수(valency)가 증가함에 따라 세포흡수는 증가되었지만, 기능적인 효과와의 상관관계는 명확하지 않다. 3가 또는 4가 접합체만이 표적세포의 luciferase 리포터 활성을 감소시켰으며, 세포흡수가 증가되는 것과 달리 50nM siRNA 농도에서 유전자 침묵효과는 포화되었다.
2.5 종양에 대한 표적 및 리간드
종양에서 과발현되는 표면 수용체는 올리고뉴클레오티드 치료제를 종양에 도입하기 위한 표적이 된다. 이상적인 표적 수용체는 정상적인 세포에서는 거의 발현이 안되고 악성세포에서만 강하게 발현되어야 한다. 또한 수용체에 대한 접근이 용이해야 하고, 종양 내에 골고루 분포되어 있어야 하며, 리간드가 결합했을 때 신속하게 세포 내 함입이 이루어져야 한다. 이러한 조건을 만족하는 수용체는 매우 적으며, 종양세포들 자체의 불균등함과 빠른 내성 획득 때문에 더욱 복잡해진다. 자주 사용되는 수용체는 Her2, 표피성장인자 수용체 EGFR, 상피세포부착분자 EpCAM 등이 있다.
엽산(folate) 수용체 또한 암세포에서 종종 발현된다. siRNA의 전달을 위해 엽산 수용체를 표적으로 한 몇 가지 보고들이 있다. Dextran과 LPEI로 구성된 나노입자 시스템을 사용하여 엽산 수용체를 표적으로 siRNA를 전달한 보고가 있었다. siRNA와 엽산 모두 PEI와 응축되기 전에 dex-tran에 공유결합으로 연결된다. In vivo에서 이 시스템은 이종이식(xenograft) 모델에서 성공적으로 GFP 발현 레벨을 감소시켰고, 종양 축적이 증가된 생체 내 분포를 나타냈다. 증가된 종양에서의 축적은 비장에서 축적되는 양이 감소하는 것과 관련이 있다. 엽산 수용체에 대해 PEI 기반의 유사한 시스템들이 in vitro, in vivo에서 평가되고 있다.
Hyaluronic acid는 glycosaminoglycan으로 상피와 신경조직 등을 포함한 다양한 조직에 널리 분포한다. 원형질막에서 생성되며 세포외기질의 주요성분을 이루고, 세포 이동, 증식, 종양성장 등에 기여한다. 이 거대 분자는 CD44의 리간드로 작용하는데, CD44는 종양에서 과발현된다. Hyaluronic acid와 양이온성 고분자 캐리어를 접합하여 종양특이적으로 siRNA를 전달하는데 사용한 보고가 있었다. 이종이식 마우스 모델에서 siRNA를 종양세포에 효율적으로 전달하였으며, 형광 리포터 유전자에 대한 침묵을 유도하였다.
Her2를 매개한 방법으로는 B형 간염 바이러스의 L 단백질(바이러스 표면 항원 단백질 HBsAg), 지질이중층, Her2 항체를 이용한 방법이 최근에 보고 되었다. In vitro에서 Her2 양성 세포에 대한 선택성과 유전자 침묵을 나타냈으며, in vivo 데이터는 아직 보고되지 않았다. 시그마 수용체의 리간드인 Anisamide가 이용되기도 하였다. 지질/protamine siRNA 나노입자의 표면에 도입된 amisamide는 세포 내 흡수를 돕는다. 이종이식 마우스 모델에서, amisamide 리간드는 추가적인 종양 축적의 증가를 나타내지는 않았다. 흥미롭게도 이 입자는 간보다 종양에서 더 높은 축적을 나타냈다. 입자크기로 인해 간에서의 유출이 방지됐을 수도 있고, 특정 이종이식 모델에서만 나타나는 현상일수도 있다. 비록, 종양으로의 전달을 증가시키지는 못했지만, anisamide를 사용한 전달 시스템은 성공적인 유전자 침묵을 일으켰다.
3가 anisamide 리간드를 스플라이싱 교정 올리고뉴클레오티드에 적용한 연구에서, 사람의 전립선암에 대한 세포 내 흡수가 증가되었고, 배양세포에서 유전자 침묵 효율 또한 4배가량 증가되었다. 신경세포와 면역세포에 높게 발현되는 cannabinoid 수용체는 anandamide 리간드를 인식한다. 이를 이용한 siRNA 접합체는 고용량이 필요하기는 했지만, 배양세포에 성공적으로 흡수되었고, 목표유전자의 침묵효과를 나타냈다.
펩타이드들도 올리고뉴클레오티드에 쉽게 부착될 수 있다. G단백질 연결 수용체(G protein coupled receptor, GPCR) BB2는 많은 악성종양세포에서 발현된다. BB2 수용체에 선택적인 히스티딘이 풍부한 펩타이드를 올리고뉴클레오티드에 결합하여, 사람 전립선암 세포에서 성공적인 스플라이싱 교정을 유도하였다. 하지만 약리학적 효과는 크지 않아서, 실제 치료에 적용할 수 있을지는 미지수이다.
Protamine과 같은 염기성 펩타이드와 융합한 항체를 이용한 전략도 있다. 양이온성 펩타이드에 Fab 단편을 융합시킴으로써 전하를 통한 siRNA와의 복합체 형성을 가능하게 한다. 항원 의존적인 조직 특이적 유전자 침묵 효과가 in vivo에서 확인되었다. 이종이식 마우스 모델에서 HIV 바이러스의 gp160 표면단백질에 대한 Fab 단편 융합을 사용하여, siRNA의 선택적인 전달과 효과를 나타낸 결과가 있었다. 유사하게, integrin에 대한 단일가닥 항체 단편을 이용한 경우나, 조류 인플루엔자 바이러스의 hemagluttinin 항원에 대한 항체 단편을 이용할 경우가 있었다. 융합 단백질은 단분자이지만, 양이온성 펩타이드와 핵산물질 사이의 전하 복합체를 형성해서 입자와 같은 구조를 형성한다. 때문에 이들은 착물 입자와 유사한 성질을 나타내며, 높은 양전하 밀도 때문에 낮은 용해도를 나타낸다. 항체 단편 대신 단백질 결합이 용이하고, 생산이 쉽고, 높은 안정성을 나타내며, 변형이 쉬운 스캐폴드를 이용하기도 한다. Designed Ankyrin Repeat protein fusion protein (DARPin)과 protamine을 이용한 시스템은 항원 양성 세포에 특이적인 유전자 침묵을 유도하였다.
자가다중화 단백질의 재조합 기술로 proteinticle이라 부르는 입자를 만들 수 있다. 세포침투 펩타이드, 표적지향 펩타이드, 올리고뉴클레오티드 결합 도메인으로 이뤄진 융합단백질이 종양 특이적인 siRNA의 전달에 사용되었다. EGFR, vimentin 표적 펩타이드의 사용은 in vitro에서 종양세포 내 세포 흡수와 유전자 침묵을 나타냈다. Proteinticle 자체는 12nm 정도의 균일한 입자크기를 갖지만, siRNA와 복합체를 이루면서 크고 불균일한 입자형태로 응집된다. 핵산결합도메인이 표면에 위치하기 때문에 핵산물질은 작은 proteinticle의 다중화를 유도한다. 결과적으로 표적지향 펩타이드는 응집체 내부에 은폐되어 수용체결합에 사용될 수 없게 된다. In vivo에서 proteinticle의 장점은 아직 밝혀지지 않았다.
항체-약물 접합체는 의약개발을 위한 매력적인 분자이지만, siRNA의 표적전달에서 보고된 바는 없다. 최근 Genentech 사는 siRNA-항체 접합체에 대한 분석 방법론을 개발하여 보고했지만, 약리학적 데이터는 아직 없다.
이 분야에서의 논문이 적은 것은, 이러한 화합물에서 siRNA의 세포 내 흡수와 세포 내 움직임에 대한 불명확함과 위치 특이적인 접합 생성이 복잡하기 때문일 것이다. 또 다른 단백질 스캐폴드는 변형과, 접합체 생성이 쉽다. 올리고뉴클레오티드는 affibody, anticalin, DARPin과 같은 스캐폴드에 위치특이적으로 접합될 수 있다. 미래에는 이러한 접합체가 다양한 단백질-수용체 상호작용과, siRNA 매개 유전자 침묵을 위해 활용될 것이다.
2.6 나노입자 약물 전달 시스템
거대분자 전달 시스템은 핵산물질의 캐리어로써 다양한 멀티컴포넌트 거대분자 전달 시스템이 개발되고 평가되었다(그림 1). 지질입자, 양이온성 고분자(PEI, chitosan, polyamine 등), 양이온성 펩타이드 등이 주류를 이룬다. 구성요소의 구조, 조성, 캡슐화 또는 복합체 형성 방법 및 비율 등의 파라미터가 조절하여 최적화함으로써 이들의 효과를 증가시키고 독성을 감소시킬 수 있었다. 원래 상태의 이러한 시스템은 수동적인 타겟팅에 의존하여 전달된다. 앞서 언급한 바와 마찬가지로, 입자 크기와 표면 조성에 따라 불가피하게 높은 신장, 간, 비장 축적을 나타내며, 다른 조직이나 기관으로는 극히 낮은 농도로 존재한다. 어떤 경우에는 EPR효과가 종양 내 축적을 촉진한다.
그림 1. 표적능 siRNA 나노입자의 여러 가지 방법에 대한 도식. 올리고뉴클레오티드는 양전하를 띤 고분자중합체, 지질과 복합체를 이루거나, 중성의 리포좀, 나노입자 속에 캡슐화된다. 복합체는 추가로 지질막으로 캡슐화될 수도 있다. 표적에 전달되기 위한 리간드는 반응성 표면이나 지질막의 변형된 구성성분을 이용하거나, 또는 조립 후 생체접합(bioconjugation)에 의해 표면에 이식된다. 종종 PEG화(PEGylation)는 비특이적 상호작용을 감소시키기 위해 사용된다.
리간드가 첨가되지 않은 중합체 중 일부는 간 이외의 다른 기관으로 상당한 전달을 나타내기도 하였다. PEI는 유전자 및 올리고뉴클레오티드 전달을 위한 캐리어로써 오랫동안 연구되었다. PEI와 핵산물질은 정전기적 상호작용에 의해 복합체를 이루며 응축된 입자를 이룬다. PEI는 길이와, 분기정도, 변형의 존재 등에 따라 다양한 고분자이다. 일반적으로 고분자의 분자량이 증가할수록 우수한 세포 내 흡수를 나타내지만, 독성도 높다. 최근, epoxide 변형된 pentadecane과 PEI600 아민을 14:1로 반응시켜 지질중합체 하이브리드가 생성되었고, 조직에서의 약리학적 효과가 평가되었다. siRNA와 고분자, PEG2000-C14가 응축되어 만들어진 입자는 마우스 모델에서 간세포 유전자 발현에는 영향을 주지 않고, 혈관내피세포에서 유전자 침묵에 효과가 있는 것으로 나타났다. 비슷하게 PEI가 포함된 입자를 사용했을 때와 다르게 예상치 않게 효율적이었다. 내피세포를 표적하는 것에 대한 분자적인 이유는 모르지만, 혈청 단백질과의 독특한 상호작용 프로파일에 의해 나타날 수 있다고 추측하고 있다.
이러한 일부 연구결과에도 불구하고, 간에 축적되는 것을 막기 위해 리간드를 사용하는 것이 필수적으로 보인다. 이는 전달시스템을 더 복잡하게 만든다. 리간드는 전달시스템의 각 구성요소에 부착될 수 있다. 예를 들면, 지질성분 중 하나에 부착하여 리포좀을 이루도록 할 수 있다. 이런 방법은, 재현성있게 리간드-캐리어 생체접합체를 얻을 수 있지만, 입자의 형성에 관한 물리화학적 파라미터들이 달라질 수 있고, 입자의 표면에 적절하게 노출되지 않아 수용체 결합 능력이 감소될 수도 있다.
표적능을 갖게 하는 분자는 올리고뉴클레오티드 캐리어가 형성된 후에, 표면의 반응성 작용기에 추가될 수도 있다. Thiol-maleimide, amide-active ester 커플링, click chemistry alkin-azide cycloaddition 등 많은 수의 부착 방법이 가능하다. 이러한 전략은 간단한 반면, 입자표면의 반응기와 부착반응의 효율에 따라서 리간드의 밀도가 달라질 수 있다. 수용체의 결합과 세포 내 함입에 충분하지 않은 리간드 밀도로 다음 단계로 진행하기 힘들 때가 종종 있다. 대부분의 경우, 전달입자의 외부표면에 여러 개의 리간드가 제시될 것이다. 때문에 입자의 결합력은 높아지고, 다시 떨어지는 속도는 낮아진다. 이는 다른 수용체(리간드에 대한 낮은 친화력을 갖는)에 결합하는 것도 가능하다는 것을 의미한다. Transferrin 수용체를 예로 들면, 높은 결합력으로 인해 라이소좀에 의한 분해가 증가되거나 미세혈관구조에 포획되는 양상을 보인다. BBB를 통과하여 뇌조직 쪽에서 떨어져 나오는 성공적인 트랜스사이토시스를 위해서 transferrin의 적절한 양이 결정적이다.
리간드의 존재가 표적세포에서 수용체 매개 엔도사이토시스를 통한 세포흡수와 저류를 증가시킬 수 있지만, 초기 조직분포에는 특이적인 영향을 미치지 않는 다는 것을 인지하는 것이 중요하다. 혈액 내 반감기와 혈관 밖으로 나와 조직으로 전달되는 것은 거의 입자의 크기와 입자표면의 생리화학적 특성에 의해서만 결정된다. 나노입자는 간, 신장, 비장에서 가장 높은 비율로 발견되며, 종양조직으로의 축적은 종양 근처 혈관내피세포의 성질에 의존한다. 나노입자가 항원을 발현하는 세포에 다다를수 없다면, 의도했던 효과는 전혀 나타나지 않을 것이다. 또는 리간드를 추가하거나 해서 나노입자 표면을 기능화시키는 것은, 물리화학적 파라미터를 변화시키고 목표한 조직에서의 축적 감소를 일으킬 수도 있다.
표적능을 가진 나노입자가 종종 예기치 않게 낮은 성능을 보이는 것은, 생체분자 코로나(biomolecular corona)라 불리는 입자표면 주위의 혈청생체분자의 접착 때문일 수도 있다. 초기 생체분자 코로나는 거의 단일분자층 형태를 이루는데, 나노입자의 성질과 주변 특정 생체분자의 양에 따라 코로나의 조성도 달라진다. 단백질들로 구성되지만 지질과 같은 다른 분자들도 존재한다. 초기 코로나층 위로 일정한 분자교환이 일어나고 있는 더 동적인 2차 코로나층이 생성되는 것으로 여겨진다. 생체분자 코로나는 나노입자의 성질을 주변 환경에 따라 변화시키게 된다. 부착된 생체분자는 약동학적인 특성에도 영향을 미칠 수 있고, 리간드의 적절한 결합을 방해할 수 있다. 표면의 PEG화(PEGylation)는 일반적으로 생체분자 코로나의 형성을 감소시켜서, 본래의 나노입자의 성질을 유지시키는데 중요하다.
2.7 표적지향 약물전달을 위한 생체접합체(Bioconjugate)
siRNA 접합체는 단일 분자로써 나노입자에 패키징된 것과는 매우 다른 성질을 갖는다. 신속한 분해를 피하기 위해서 siRNA 구성요소는 2’-O-methylation, 2’-fluoridation 등의 화학적 변형이 필요하다. 부착된 리간드의 크기에 따라서 신장 배설이 위협이 될 수도 있다. 이는 PEG화(PEGylation)와 같은 방법으로 분자 크기를 키움으로써 막을 수 있다. 입자를 이용한 방법과 다르게 생체접합체는 혈관내피세포의 구멍 등에 의존하지 않기 때문에 기관으로의 분포는 쉽게 이뤄진다. 간에서의 흡수와 축적을 더욱 증가시키기 위해서 지질, 스테로이드, 토코페롤, 탄수화물과 같은 리간드가 사용되었다(그림 2). GalNAc 플랫폼의 성공을 고려할 때, 다른 기관에서의 흡수와 축적을 증가시키는 방법이 개발되지 않은 것이 놀랍다.
소분자 리간드의 경우 올리고뉴클레오티드를 합성할 때 삽입된 적절한 링커를 통해서, 상대적으로 쉽게 siRNA의 센스 또는 안티센스 가닥에의 3’, 5’ 말단에 접합될 수 있다. 부착 방법은 리간드에 따라 선택되어 사용될 수 있다. 펩타이드 접합체의 경우, 올리고뉴클레오티드와 펩타이드가 각각 따로 합성된 후 연결하거나, 또는 단계적인 고체상 합성(solid-phase synthesis)을 이용할 수 있다. 펩타이드의 길이가 길어질수록 보호기 등이 요구되는 고체상 합성 방법은 더 복잡해진다.
RNAi 복합체를 활성화시키기 위해서는 siRNA가 캐리어로부터 유리되어야만 한다. 따라서 엔도좀의 산성 환경에서는 절단 될 수 있도록 고안된 불안정한 결합이 유리한 것으로 간주된다. 하지만 이러한 과정이 세포 내에서 어떻게 일어나는지 정확하게 알려지지 않았기 때문에, 표적능을 위한 리간드가 반드시 절단될 필요가 있는지는 불명확하다. 또한 캐리어 자체에 의존적일 수도 있다. 예를 들어, 3가 GalNAC 접합체는 안정적인 결합에도 불구하고 효율적인 유전자 침묵을 전혀 방해하지 않았다.
다가 결합은 여러 개의 표면 수용체에 동시에 결합할 수 있기 때문에 세포결합과 흡수능력이 종종 우수하게 나타난다. 이를 위해서는 리간드 간의 거리 및 공간적 배열이 중요하다.
그림 2. 생체접합체 기술 및 리간드 구조의 개관. 올리고뉴클레오티드에서의 부착지점은 주로 3’- 및 5’- 말단의 수산화기가 있는 곳이 된다. 가장 많이 쓰는 결합유형은 엔도좀에서 분해될 수 있는 불안정한 링커인 이황화결합 및 안정적인 연결을 위한 click 화학에서 유래한 maleimide 또는 triazole 결합이다.
3. 결론
siRNA 전달 시스템에 대한 10여년의 연구개발은 중요한 파라미터가 무엇인지 등 많은 교훈을 남겼다. 나노입자의 효능은 그 크기에 크게 의존하고, 입자의 재료와 표면의 특성도 중요하다. 최적화된 입자크기는 신속한 배설(작은 입자의 경우)을 방지하고, 세망내피계로부터 제거(큰 입자의 경우)를 피하기 위해 중요하다. 입자 재료에 따라 다르지만, 최적의 크기는 약 50-200nm로 확립되었다. 다공성은 물의 침투와, 그에 따른 방출 및 분해에 중요한 쟁점이 된다. 표면의 PEG화(PEGylation)는 입자가 면역세포에 포획되어 면역계를 활성화 시키는 것을 방지하지만, 일반적으로 세포흡수와 세포 내 유리를 감소시킨다. 양전하는 세포와의 결합을 증가시키고, pH 완충능력을 통해 엔도좀 시스템을 붕괴시킴으로써 세포 내 전달을 증가시키는 것으로 생각되지만, 면역 세포와의 비특이적인 상호작용을 증가시켜 독성을 유발하기도 한다.
이러한 성질들은 나노캐리어를 디자인하는데 도움이 되지만 모든 곳에서 적용되지는 않는다. 면역원성 및 독성 효과는 대게 예측할 수 없으며, 기능을 위한 성공적인 전달 정도는 종종 놀랄 만큼 낮다. 좀더 합리적인 디자인을 위해서는 정확한 세포 내 흡수과정과, 세포 내 움직임, 엔도좀 분류 구분 등에 관한 더 많은 지식이 필요하다. 수용체 매개 흡수를 이용할 때는 고친화력 리간드를 이용한 생체접합체를 통해 표적 조직이나 기관 축적이나 특이적인 세포와의 결합을 증가시킬 수 있다. 나노캐리어 입자와 유사하게, 세포질 내로 전달되는 정도는 엔도좀탈출과 엔도좀 분류 구분에 크게 의존한다. 라이소좀 분해정도와 기능적인 흡수를 증가시키기 위한 전략을 찾고, 최적의 수용체와 그에 상응하는 리간드를 찾기 위해서는 더 자세한 연구들이 필요하다.
학술적 연구와 실제 치료 적용 사이의 괴리도 존재한다. 학술적 나노기술 연구는 흡수 기전에 대한 최신의 식견을 얻기 위해서 복잡한 최첨단시스템을 개발하는 경우가 많다. 이러한 정교한 시스템은 제작방법, 특성화 방법, 독성 등의 문제로 인해 실제 임상환경에 적용될 가능성은 거의 없다. 좀더 단순화된 해결방법이 doxorubicine이나 paclitaxel 약물요법을 향상시키고 시장 승인을 따냈지만, 생체거대분자의 전달에도 적용될 가능성은 적다.
***siRNA 전달의 경우, 두 가지 방법이 성공적으로 전임상단계와 조기임상단계의 시험을 받는 중이다. SNALPs (lipid particles designed for nucleic acid, 핵산을 위해 설계된 지질분자) 캡슐 내에 담는 방법과 asialoglycoprotein 수용체 리간드인 N-acetyl galactosamine 접합체를 이용한 방법이 있으며, 두 가지 방법 모두 간을 표적으로 한다. 여기에서부터 출발하여 수용체 리간드를 전환하는 방법으로 siRNA 요법을 다른 장기로 확장시킬 수 있을 것이다. 지질입자를 진보시키는 것은 더욱 복잡하다. 지질친화성 및 입자크기 모두에 의해서 간 축적이 유발되기 때문이다. 표면의 특성은 구조적 변형(PEGylation, 타겟팅 부위의 부착)을 통해 조절될 수 있지만, 입자의 크기 때문에 치밀한 내피세포접합을 갖는 조직은 모두 배제되고 여전히 간으로 축적을 일으킬 것이다. 더 효과적인 나노입자의 표적화 전략이 개발되면 획기적인 발전이 이뤄질 수 있을 것이다.
siRNA 전달 시스템에 대한 세포 내 흡수와 세포 내 움직임에 대한 기초적인 기전의 이해와 구조적 및 생물리학적 요구조건에 대한 이해가 증진됨에 따라 가까운 미래에 이 분야에서의 획기적인 발전이 예상된다. 향후 10년 동안의 임상현장으로의 성공적인 적용과 그에 따른 임상적 결과는 siRNA 치료법의 앞으로의 파장을 결정할 것이다.
US regulators have approved the first therapy based on RNA interference (RNAi), a technique that can be used to silence specific genes linked to disease. The drug, patisiran, targets a rare condition that can impair heart and nerve function.
The approval, announced by the US Food and Drug Administration on 10 August, is a landmark for a field that has struggled for nearly two decades to prove its worth in the clinic. Researchers first discovered RNAi 20 years ago1, sparking hopes of a revolutionary new approach to medicine. Since then, however, a series of setbacks has lessened those expectations.
“This approval is key for the RNAi field,” says James Cardia, head of business development at RXi Pharmaceuticals in Marlborough, Massachusetts, which is developing RNAi treatments. “This is transformational.”
Patisiran works by silencing the gene that underlies a rare disease called hereditary transthyretin amyloidosis. In that illness, mutated forms of the protein transthyretin accumulate in the body, sometimes impairing heart and nerve function.
The drug's approval means that pharmacology textbooks will need to be rewritten, says Ricardo Titze-de-Almeida, who studies RNAi at the University of Brasilia. “We are inaugurating a new pharmacological group,” he says. “We will have many more such drugs in the coming years.”
This was the hope when Alnylam, the company in Cambridge, Massachusetts, that developed patisiran, launched in 2002. Four years later, theNobel Prize in Physiology or Medicine was awarded to two RNAi pioneers: Andrew Fire of Stanford University School of Medicine in California and Craig Mello of the University of Massachusetts Medical School in Worcester.
But to make RNAi into medicine, developers would first need to determinehow to deliver delicate molecules of RNA safelyto their target organs. They needed a way to shield the RNA from degradation in the bloodstream, prevent it from being filtered out by the kidneys, and allow it to exit blood vessels and spread through tissues. “That proved to be a substantially harder problem than we anticipated,” says Douglas Fambrough, chief executive of Dicerna, an RNAi-focused company in Cambridge, Massachusetts.
As researchers grappled with the delivery puzzle, investors began to lose confidence. In 2008, analyst Edward Tenthoff of investment bank Piper Jaffray in New York City advised his clients to stop buying Alnylam stock. “We saw the promise in the technology, but the delivery was lacking,” he says.
But gradually, some RNAi companies began to iron out the kinks in their delivery systems, and Tenthoff started to encourage investors to buy stock again. Alnylam experimented with a number of delivery routes and target organs, encasing some of its RNA molecules in fatty nanoparticles orchemically modifying the RNAs to help them survive the perilous journey through the bloodstream.
RNAs protected in this way and injected into the bloodstream tended to accumulate in the kidneys and liver. This led the company to look at transthyretin, which is produced mainly in the liver. In a clinical trial in 225 people with hereditary transthyretin amyloidosis who showed signs of nerve damage, average walking speed significantly improved in those who received the treatment2. Walking speed declined in the placebo group.
In the future, Alnylam and others will be able to move beyond the liver, says company co-founder Thomas Tuschl, a biochemist at Rockefeller University in New York City. Quark Pharmaceuticals of Fremont, California is testing RNAi therapies that target proteins in the kidneys and the eye. Alnylam is developing ways to target the brain and spinal cord, and Arrowhead Pharmaceuticals of Pasadena, California, is working on an inhalable RNAi treatment for cystic fibrosis.
“I’ve never been more optimistic about the future of RNAi,” says Fambrough. “All of those tear-your-hair-out days were worth it to get to today.”
Advances in RNA delivery might also benefit researchers who are developing gene-editing therapies based on the popular technique CRISPR–Cas9. That system uses a DNA-cutting protein called Cas9, which is guided to the desired site in the genome by an RNA molecule.
Like RNAi before it, CRISPR–Cas9 has become a common tool in genetics laboratories. But it might still face a difficult and lengthy path to the clinic. Much like ordinary drugs, RNAi therapies will break down over time; a gene edit, however, is intended to be permanent, which amplifies concerns about safety.
“I hope they can do it more quickly than we did it, but I would not expect it to be so smooth,” says Fambrough. “I wish them the best of luck.”
미국규제당국은질병과관련된특정유전자를침묵시키는데사용될 수 있는 기술인RNA간섭(RNAi)에 기초한 첫번째치료법을승인했다. 이 약인파티시란은심장과신경기능을손상시킬 수 있는 희귀한상태를목표로 하고 있다. 8월10일미국식품의약국이발표한 이 승인은 이 클리닉에서그것의가치를증명하기 위해 거의20년동안고군분투해 온 분야의랜드마크다. 연구원들은20년전에처음으로RNAi를발견했는데, 이는의학에대한혁명적인새로운접근에대한희망을불러일으켰다. 그러나 그 이후로일련의좌절은그러한기대를감소시켰다. RNAi치료를개발하고 있는 매사추세츠주말버러에 있는 RXi제약회사의제임스카디아사업개발책임자는 "이번승인은RNAi분야의핵심"이라고 말한다. “이건 변혁적인 거야.”
Patisiran은유전성트랜스티레틴아밀로이드증이라고불리는희귀한질병의기초가 되는 유전자를침묵시킴으로써작용한다. 그 질병에서, 돌연변이된형태의단백질트랜스티레틴은신체에축적되어때로는심장과신경기능을손상시킵니다. 브라실리아대학에서RNAi를연구하는리카르도티츠데알메이다는 이 약의승인은약리교과서를다시써야 한다는 것을 의미한다고말한다. 그는 "우리는새로운약리학그룹을출범시키고 있다"고 말했다. "앞으로몇 년 안에그런약들을더많이갖게 될 겁니다."
2002년패티시란을 개발한 매사추세츠주캠브리지에 있는 알닐람이출범했을때희망이었다. 4년 후, 노벨생리학또는의학상은캘리포니아의스탠포드 의과 대학의 앤드류파이어와우스터의매사추세츠 의과 대학의 크레이그멜로라는두명의RNAi개척자에게수여되었습니다.
그러나RNAi를의학으로만들기 위해서는 개발자들이먼저RNA의섬세한분자를그들의목표장기에안전하게전달하는방법을결정할필요가있을 것이다. 그들은RNA가혈류의저하로부터보호하고신장에의해여과되는 것을 방지하고혈관을빠져나와조직을통해퍼지도록하는방법이필요했습니다. 매사추세츠주캠브리지에 있는 RNAi중심회사인디세르나의더글러스팜브러사장은 "이는 우리가예상했던것보다훨씬더어려운문제임이 입증됐다"고 말했다. 연구자들이배달퍼즐을고심하면서투자자들은자신감을잃기 시작했다. 2008년뉴욕의투자은행파이퍼재프레이의분석가에드워드텐토프는고객들에게알닐람주식구매를중단하라고조언했다. 그는 "기술에서약속을봤지만배달이부족했다"고 말했다.
2010년까지대형제약회사들도RNAi에대한욕구를 잃고, 협업을끊고, 내부연구프로그램을끝내고있었다. “대부분큰약국에서죽은RNAi를남겼습니다.” 팜브러가말한다. 안전에 대한 우려는2016년알닐람이임상실험에서환자사망과의연관성을발견한후주요RNAi프로그램중하나를포기하면서다시한번타격을 입혔다.
그러나점차적으로일부RNAi회사들은배달시스템의꼬임을해결하기시작했고, 텐토프는투자자들에게다시주식을사도록장려하기 시작했다. 알닐람은많은전달경로와표적장기를실험하여지방나노입자에RNA분자의일부를담가RNA를화학적으로수정하여혈류를통한위험한여정에서살아남도록 도왔다. 이런식으로보호되고혈류에주입된RNA는신장과간에서축적되는 경향이 있었다. 이로 인해회사는주로간에서생산되는트랜스티레틴을보게되었습니다. 신경손상의징후를보인유전성트랜스티레틴아밀로이드증환자225명을대상으로한임상실험에서치료받은환자에서평균보행속도가유의하게향상되었다.2. 위약군에서보행속도가감소했다.
앞으로알닐람등은간을 넘어설 수 있을 것이라고 뉴욕시록펠러대학의생화학자인토마스투슐공동창업자는말한다. 캘리포니아주프레몬트의쿼크제약회사는신장과눈의단백질을목표로 하는 RNAi치료법을시험하고 있다. 알닐람은뇌와척수를목표로하는방법을개발하고있으며, 캘리포니아주패서디나의애로우헤드제약회사들은낭포성섬유증에대한흡입가능한RNAi치료를연구하고 있다. 팜브러는 "RNAi의미래에대해 이보다 더낙관적인 적이 없다"고 말한다. “그머리털이찢어지는날들은오늘까지 갈 만한 가치가 있었어.” RNA전달의진보는또한인기 있는 기술인CRISPR-Cas9에 근거하여 유전자편집치료법을개발하고 있는 연구자들에게도도움이 될 수 있다. 이 시스템은RNA분자에 의해 게놈의원하는부위로안내되는Cas9라고불리는DNA절단단백질을사용한다. CRISPR-Cas9는이전의RNAi처럼유전학실험실에서흔히 볼 수 있는 도구가 되었다. 하지만그것은여전히클리닉으로가는어렵고긴길에직면할수도 있다. 일반약물과마찬가지로RNAi치료는시간이 지남에 따라 분해 될 것입니다. 그러나유전자편집은영구적이어야하며안전에 대한 우려를증폭시킵니다. “우리보다더빨리할 수 있으면 좋겠지만, 그렇게순조롭다고는기대하지않을 겁니다.”
Hereditary transthyretin amyloidosis is an autosomal dominant, multisystemic, progressive, life-threatening disease caused by mutations in the gene encoding transthyretin (TTR).1The liver is the primary source of circulating tetrameric transthyretin protein. In hereditary transthyretin amyloidosis, both mutant and wild-type transthyretin deposit as amyloid in peripheral nerves and the heart, kidney, and gastrointestinal tract,1,2resulting in polyneuropathy and cardiomyopathy.1,3Neuropathic changes result in profound sensorimotor disturbances, with deterioration in activities of daily living and ambulation.4Autonomic nerve involvement causes hypotension, diarrhea, impotence, and bladder disturbances.4Cardiac manifestations include heart failure, arrhythmias, orthostatic hypotension, or sudden death due to severe conduction disorders.5Hereditary transthyretin amyloidosis is inexorably progressive, with survival of 2 to 15 years after the onset of neuropathy6-8but only 2 to 5 years among patients presenting with cardiomyopathy.9,10
Current treatment options for hereditary transthyretin amyloidosis are limited and include orthotopic liver transplantation and transthyretin tetramer stabilizers (tafamidis or diflunisal). However, many patients who are treated with these approaches continue to have disease progression.4,10-16
RNA interference (RNAi) is an endogenous mechanism for controlling gene expression. It results in the cleavage of target messenger RNA (mRNA) by small interfering RNAs bound to the RNA-induced silencing complex. Patisiran, a hepatically directed investigational RNAi therapeutic agent (Fig. S1 in theSupplementary Appendix, available with the full text of this article at NEJM.org), harnesses this process to reduce the production of mutant and wild-type transthyretin by targeting the 3′ untranslated region of transthyretin mRNA.17Previously, dose-dependent reduction of circulating transthyretin levels has been observed with patisiran administration in healthy volunteers and in patients with hereditary transthyretin amyloidosis.17,18In addition, patisiran has shown the potential to halt the disease or improve disease control in a phase 2, open-label extension study involving patients with hereditary transthyretin amyloidosis.19Here we present efficacy and safety data from the APOLLO trial, a randomized, placebo-controlled, phase 3 trial involving patients with hereditary transthyretin amyloidosis with polyneuropathy.
유전성트랜스티레틴아밀로이드증은트랜스티레틴(TTR)을 암호화하는유전자의돌연변이에의해야기되는상염색체우성, 다체계, 진행성, 생명을위협하는질병이다.1간은순환하는테트라메릭트랜스티레틴단백질의주요공급원이다. 유전성트랜스티레틴아밀로이드증에서는말초신경과심장, 신장, 위장관에돌연변이와야생형트랜스티레틴이아밀로이드로침전되어1,2가다신경병증과심근병증을일으킨다.1,3신경병증의변화는심오한감각운동장애를일으키며일상생활과아밀로이드의활동이악화된다.4자율신경관여는심오한감각운동장애를일으킨다. 저혈압, 설사, 발기부전, 방광장애.4심장발현에는심부전, 부정맥, 정형외과저혈압, 또는심한전도장애로인한갑작스런사망이 포함된다.5유전성트랜스티레틴아밀로이드증은신경병증6-8이발병한지2년에서15년이지난 후, 심장근육을보이는환자중2년에서5년밖에생존하지못한다. 병리학9,10 유전성트랜스티레틴아밀로이드증에 대한 현재의치료옵션은제한되어있으며, 정형외과간이식및트랜스티레틴테트라머안정제(타파미디스 또는 디플루니살)를 포함한다. 그러나이러한접근법으로치료를받는많은환자들은계속해서질병진행을하고 있다.4,10-16
RNA간섭(RNAi)은 유전자발현을조절하는내생적메커니즘이다. 이는RNA에의해유도된침묵복합체에결합된작은간섭RNA에 의해 표적메신저RNA(mRNA)의 분열을 초래한다.패티시란, 간지시적으로지시된조사RNAi치료제(그림).본논문의전체텍스트와함께NEJM.org에서사용 가능한 보충부록의S1은트랜스티레틴mRNA의3개의번역되지 않은 영역을목표로돌연변이및야생형트랜스티레틴의생산을줄이기 위해 이 과정을활용한다.17이전에는건강한자원봉사자와유전성트랜스티레틴환자에서패티시란투여로순환하는트랜스티레틴수준의선량의존적감소가관찰되었다. 또한, 패티시란은유전성트랜스티레틴아밀로이드증환자를대상으로하는2단계개방표지확장 연구와 함께질병을멈추거나질병을억제할 수 있는 가능성을보여주었다.19여기서우리는무작위위약조절, 3단계임상시험인APOLLO시험에서효능감과안전데이터를제시한다. 요로병증
아직까지 상업화에 성공한 RNAi 치료제는 없지만 임상 막바지 단계를 거치고 있는 이 분야가 한번 시작되면 큰 파도가 일 것이라고 보는 전문가들이 적지 않다.
한국분자·세포생물학회의 'RNAi 치료제 최근 연구 동향'에 따르면, `RNA 간섭(RNA interference, RNAi)` 현상은 이중가닥의 RNA(dsRNA)를 세포 안으로 집어 넣어주면, Dicer라는 리보핵산 가수분해효소에 의해 잘려 21-23bp의 작은 RNA로 전환되는 과정이다. 여기서 잘린 작은 RNA 형태를 `siRNA(short interfering RNA)`라고 부른다.
이 RNAi 분야는 실질적으로 모든 유전자의 발현을 억제 시킬 수 있으므로 antisense, ribozymes 보다 훨씬 더 큰 잠재력을 가진 것으로 평가된다. 따라서 최근의 연구동향은 마이크로 RNA의 서열 특이적인 유전자를 억제시키는 siRNA를 표적세포에 전달시켜 다양한 질환에 적용하는 추세다.
RNAi 치료제는 모든 유전자를 특이적으로 공략하기 때문에 치료제가 없는 난치질환에 새로운 해결책으로 부상하고 있으며, 다양한 질병에 적용이 가능하다는 점에서 차세대 미래 신약 기술로 인정받고 있다.
하지만 이런 장점에도 불구하고, RNAi 치료제가 신약으로 실현되기 위해서는 여러 가지 문제점들을 극복해야 한다. 우선 siRNA 자체의 화학적 변형이나 구조적 안정화, 전달체 개발을 통해 생체 내 안정성을 해결해야만 한다.
또한 나노 입자의 사이즈, 표면 전하, 형태를 조절해 전달 효율을 증가시키는 것이 중요한 사안으로 여겨진다. 이러한 조건들을 충족시킬 때, 보다 안정적이면서 긴 시간 동안 특정 조직에 siRNA를 정확하게 전달할 수 있다.
다행인 점은 RNAi 치료제의 단점을 보완한 `siRNA 전달체`의 개발로 전신 투여를 위한 많은 수의 약물이 등장했다는 것이다. 2004년 처음으로 siRNA를 이용한 RNAi 치료제가 임상시험을 시작한 이 후로, 20종 이상의 siRNA 치료제가 개발되고 있다.
<임상단계의 siRNA 치료제,RNAi 치료제 최근 연구 동향>
물론 그 과정이 쉽지만은 않았다. 'Bevasiranib'은 임상 3상에서 낮은 치료 효과로 중단됐고, AGN-745 또한 off-target effect 로 임상 2상 단계에서 실험을 중단했다. TKM–ApoB는 임상 1상에서 콜레스테롤의 일시적인 감소현상이 나타나 임상이 종결된 경우다. 이들 siRNA치료제는 투여했을 때 공통적으로 TLR(Toll Like Receptor)를 활성화 시켜 면역 부작용을 일으켰다.
반면 Quark Phama의 'I5NP'는 다른 naked siRNA약물과 달리 정맥주사 시 임상 2상까지 괄목할 만한 치료 효과를 보여줬다.
제약업계가 주목하는 `siRNA 전달체 플랫폼`은 많은 선행 연구를 통해 리포좀을 기반으로 한 전달체가 보다 안정적으로 치료제의 효과를 이끈다는 보고에 영향을 받았다. Alnylam/Tekmira의 Nucleic acid lipid particle(SNALP)를 기반으로 한 'ALNVSP02' 치료제는 임상 1상을 마쳤으며, 일반적으로 간암 환자 의 50%가 항-VEGF 효과를 보여줬다.
SNALP 치료제인 Alnylam의 'ALN-TTR01'은 transthyretin(TTR) 단백질이 감소되는 효과를 보였고, 또다른 ATTR siRNA 치료제인 'ALN-TTR02'는 단 한번 의 투여로 TTR 단백질이 94프로까지 억제되는 것을 보여줬다.
이외에 괄목할 만한 RNAi 치료제인 Silence Therapeutics의 'Atu027'은 양이온 리포좀인 AtuPLEX와 결합된 복합체로 임상 1상에서 독성 실험 결과 33명의 암환자 중 27명에서 부작용이 발생하지 않았다.
국내에서는 유한양행과 함께 면역 항암제 공동 개발에 나선 바이오니아가 고효율의 생분해성 자가조립 나노입자인 SAMiRNA 기술을 바탕으로 siRNA 핵심 신약과 신약후보 물질을 개발하고 있다.
이 기술은 RNAi 기술의 단점으로 작용하던 off-target effect를 줄이고 간 독성 역시 현저하게 낮은 것으로 나타났다. 현재 여러 차례의 동물 실험을 통해 SAMiRNA 나노 입자를 사용한 종양 크기 성장의 억제가 효과적임을 확인했고 임상시험을 준비 중이다.
올릭스 역시 RNAi 기반의 핵산 약물을 개발하고 있는 바이오텍 회사이다. 올릭스는 기존 siRNA의 면역 반응 유발이나 비 표적 유전자 억제와 같은 부작용을 감소시킨 비대칭 자가전달 RNAi 기술과 전달체 없이 원하는 세포 내로 RNA를 전달하는 자가전달 RNAi 기술의 원천 특허를 보유하고 있다.
이러한 원천 기술을 바탕으로 올릭스는 비대 흉터 치료제 `OLX101`의 비임상 시험을 마치고 국내는 물론 아시아 최초 RNAi 제품의 임상단계에 진입했다. 이와 동시에 하반기부터 유럽에서 1상 임상시험을 준비하고 있다.
아울러 올릭스의 특발성 폐섬유화 치료제 `OLX-201` 개발은 싱가포르와 공동 진행하고 있으며, 황반변성 치료제인 `OLX301`의 비임상시험을 진행 중이다. 두 제품 모두 내년 중 해외 임상을 준비하고 있다.
인코드젠은 siRNA가 체내의 miRNA로 잘못 인식돼 발 생하는 off-target effect를 줄이기 위해 miRNA의 구조 등을 연구, 6번째 피봇(Pivot)의 위치를 변형함으로써 miRNA로의 작용을 차단하는 siRNA-6pi 기술을 고려대 연구진들과 개발했고, 그 결과를 2015년 네이처 커뮤니케이션 학술지에 게재했다. 고려대와 인코드젠은 산학협력의 형태로 기술을 이용한 다양한 질환에 대한 적용을 준비 중이다.
이처럼 siRNA 약물은 서열 특이적으로 유전자 발현을 억제시킬 수 있으므로 질병을 치료할 수 있는 무한한 가능성을 갖고 있다. 새로운 기술과 연구의 발달로 siRNA의 작용을 변화시켜 치료 가능성을 증가시켰으며 off target effect를 감소시켰다. 따라서, 현재 많은 siRNA 표적이 임상연구로 계속해서 진행 중이며 미래 신약 개발의 새로운 패러다임을 보여주고 있다.
한 업계 관계자는 "지금까지 품목허가를 받은 RNA 치료제 중 상업적으로 성공한 사례는 없다. 그러나 Ionis의 'Volanesorsen'과 같은 성공적인 임상 소식과 함께 신규 치료제 발매가 기대되고 있다. 이 RNA 치료제는 심혈관계, 퇴행성 뇌질환, 안과적 질환, 대사 질환, 바이러스 감염과 같은 질병에도 부작용을 최소화 하면서 적용할 수 있다. 앞으로 RNAi 치료제 효과를 높이기 위한 좋은 전달 시스템을 구축한다면 기존 약물치료의 패러다임을 벗어나 큰 영향력을 끼칠 것"이라고 전망했다.
매년 10월 노벨과학상 수상자가 발표되면 분야에 따라 반응이 천차만별이다. 지난해 생체시계 같은 경우는 내용이 어렵지 않고 우리가 체험하는 현상이기도 해서 관심이 높았다. 반면 어떤 경우는 그런 분야가 있었는지도 몰랐던지라 어리둥절하다.
2006년 노벨생리의학상이 그런 경우로, ‘RNA간섭(interference)’ 현상을 발견한 생물학자 두 사람이 수상했다. RNA야 DNA와 비슷한 생체분자라고 배운 것 같은데, 사람도 아닌 분자가 뭘 간섭한다는 건지 짐작도 안 된다.
사실 RNA간섭은 1998년 처음 보고된 현상으로 불과 8년 만에 선정된 것이다. 노벨상이 이렇게 빨리 주어지는 건 흔치 않은 일로 그만큼 중요한 발견이라는 뜻이다. 다만 대중에게 미처 알려지기도 전에 노벨상을 받아 이런 반응이 나온 것이다.
RNA간섭이 발견 즉시 높게 평가된 건 유전자의 발현을 조절하는 전혀 새로운 메커니즘인 데다 그 잠재력도 엄청났기 때문이다. 즉 RNA간섭으로 특정 유전자의 발현을 쉽게 조절할 수 있어 많은 아이디어를 실험으로 확인할 수 있게 됐고, RNA간섭 약물을 만들면 의학계의 혁명을 몰고 올 수도 있기 때문이다.
그러나 모든 게 예상대로 되지는 않았다. RNA간섭은 오늘날 생명과학 실험에 일상적으로 쓰이고 있지만 이를 적용한 의약품은 나오지 않고 있다. RNA간섭을 연구하는 많은 벤처에 엄청난 돈을 쏟아부었음에도 별 진전이 없는 걸로 알고 있었다.
그런데 지난 10일 미 식품의약국(FDA)은 최초의 RNA간섭 의약품을 승인한다고 발표했다. 미국의 바이오벤처 앨나이람 파마슈티컬즈(Alnylam Pharmaceuticals)의 파티시란(patisiran. 제품명은 온파트로(Onpattro))을 유전성 트랜스티레틴 아밀로이드증으로 말초신경병증을 앓는 환자의 치료에 사용할 수 있도록 허가했다. RNA간섭 현상을 발견한 지 20년 만에 마침내 RNA간섭이 의학계에 데뷔한 것이다.
현재 앨나이람은 십여 가지 다른 RNA간섭 약물 임상을 진행하고 있고 다른 여러 회사들도 수십 가지 임상을 진행하고 있다. 파티시란 승인을 계기로 머지 않아 RNA간섭 의약품이 쏟아져 나올 것으로 보인다. 아울러 RNA간섭 농약도 출시가 머지않았다. 의학과 농업에 큰 파장을 미칠 것으로 보이는 RNA간섭에 대해 알아보자.
pixabay 제공
예상치 못한 결과 나와
1953년 DNA이중나선이 밝혀진 뒤 과학자들은 DNA의 염기 서열 정보가 어떻게 단백질의 아미노산 서열로 번역되는지 밝히는 연구에 뛰어들었다. 그 결과 DNA가 먼저 같은 염기순서인 단일가닥 전령RNA(mRNA)로 전사된 뒤 리보솜에서 mRNA를 읽어 번역이 일어난다는 사실을 규명했다.
이 과정을 들여다보면 어렵지 않게 한 아이디어가 떠오른다. mRNA의 염기서열과 상보적인 서열의 RNA 단일가닥을 만들어 넣어주면 mRNA에 달라붙어 이중가닥을 만들고 결국 번역이 일어나지 못할 것이다. 아미노산의 정보, 즉 의미를 지닌 mRNA를 센스(sense)라고 부르고 여기에 상보적인 RNA를 안티센스(antisense)라고 부른다.
1980년대 이 아이디어를 확인하는 실험이 진행됐고 정말 안티센스RNA가 어느 정도 유전자 발현을 억제하는 것으로 밝혀졌다. 그러나 후속 실험들에서 결과가 들쑥날쑥했고 심지어 센스RNA(mRNA의 일부분과 서열이 같은 조각)를 넣어줘도 비슷한 효과가 나타나는 등 이해가 안 되는 결과가 이어졌다.
미국 카네기연구소의 앤드루 파이어와 매사추세츠대 크레이그 멜로는 이 혼란을 정리하기 위해 체계적인 실험을 설계해 진행했다. 즉 여러 유전자에 대해 안티센스와 센스, 그리고 둘 다를 넣어 발현 억제 정도를 비교하기로 한 것이다. 기존 이론에 따르면 안티센스RNA를 넣어준 시료에서만 효과가 있을 것이다.
그러나 누구도 예상하지 못한 결과가 나왔다. 센스는 물론 안티센스도 효과가 없거나 미미한 반면 둘을 다 넣어준 시료에서는 단백질이 거의 만들어지지 않는 강력한 효과가 나타났다. 센스와 안티센스를 같이 넣어주면 자기들끼리 먼저 달라붙어 RNA이중가닥을 만들 텐데 어떻게 이게 mRNA에 작용할 수 있을까.
더 이상한 건 세포 안에서 만들어질 mRNA보다 훨씬 적은 양을 넣어줘도 이런 효과를 보인다는 것이다. 이는 기존의 ‘1:1 대응 메커니즘’, 즉 안티센스RNA 분자가 mRNA 분자에 달라붙어 못쓰게 한다는 가정이 틀리다는 얘기다. 이를 설명하려면 넣어준 RNA가 촉매처럼 스스로는 소모되지 않은 채 상보적인 mRNA를 계속해서 파괴한다고 볼 수밖에 없다. 따라서 연구자들은 이 과정에서 뭔가 복잡한 시스템이 동원된다고 주장했다.
이 연구결과를 담은 논문은 1998년 2월 19일자 ‘네이처’에 실렸고 지금까지 1만7000회 가까이 인용된 ‘전설’이 됐다.
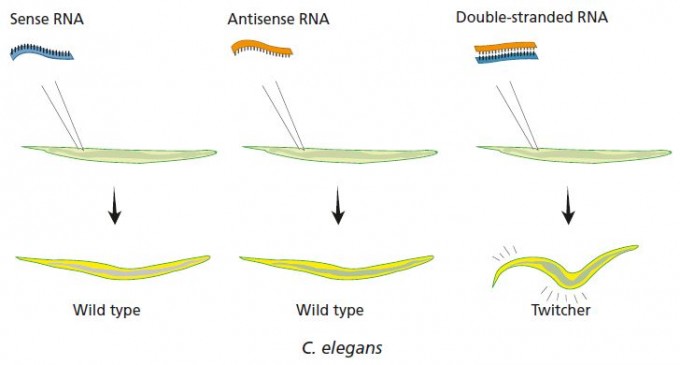
1998년 학술지 ‘네이처’에 발표한 논문에서 앤드루 파이어와 크레이그 멜로는 표적 유전자와 같은 서열인 RNA(센스)나 상보적인 서열인 RNA(안티센스)는 발현 억제 효과가 거의 없고 RNA이중가닥이 효과가 크다는 사실을 발견했다. 그림은 unc-22유전자를 대상으로 한 결과로, RNA이중가닥을 넣어 발현이 크게 떨어진 결과 예쁜꼬마선충이 경련을 일으킨다. - 노벨재단 제공
새로운 유전자 발현 조절 시스템
파이어와 멜로를 비롯해 연구자들은 후속 연구를 통해 이 시스템의 실체를 밝히는 데 성공했다. 즉 세포 안으로 RNA이중가닥이 들어오면 다이서(Dicer)라는 단백질복합체에서 염기 20여 개 길이로 잘린 뒤 또 다른 단백질복합체인 RISC에서 단일가닥(안티센스)만 남는다. 여기에 표적이 되는 mRNA가 지나가다 걸리면 이를 분해하는 반응이 일어나 mRNA가 파괴된다. 즉 짧은 RNA단일가닥이 가이드 역할을 하는 셈이다. 그렇다면 왜 이처럼 번거로운 RNA간섭 시스템이 존재하는 것일까.
파이어는 바이러스에 대한 방어시스템일 수 있다고 제안했다. 즉 RNA이중가닥 게놈을 지닌 바이러스가 침투했을 경우 다이서가 바이러스 게놈을 처리해 RISC가 안티센스 조각을 지니고 있으면 바이러스의 mRNA를 파괴해 증식을 억제할 수 있기 때문이다.
그런데 이보다 5년 앞선 1993년 발견된 마이크로RNA(miRNA) 역시 동일한 시스템을 이용해 작동한다는 사실이 밝혀졌다. miRNA는 생명체가 자신의 유전자 발현을 조절하기 위해 만들어내는 RNA간섭이었던 셈이다. 2000년대 초 RNA간섭의 실상이 드러나면서 생명과학이라는 조각퍼즐의 상당 부분이 한꺼번에 맞춰졌기 때문에 2006년 서둘러 노벨상이 주어진 것이다.
RNA간섭은 생명과학 실험에 큰 영향을 미쳤다. 즉 어렵지 않게 특정 유전자의 발현을 80~90%를 억제할 수 있게 되면서(이를 유전자 녹다운(knockdown)이라고 부른다) 그 유전자의 기능을 규명할 수 있게 됐기 때문이다. 특히 유전자를 아예 고장 내면(이를 녹아웃(knockout)이라고 부른다) 생물이 발생과정에서 죽을 경우 그 기능을 알 방법이 없는데(무척 중요할 것임에도), 녹다운이 큰 도움이 됐다.
2000년대 초 RNA간섭의 대략적인 작동 메커니즘이 밝혀졌다. 이에 따르면 RNA이중가닥에서 mRNA에 상보적인 단일가닥이 RISC에 결합돼 RISC가 표적인 mRNA를 파괴하는데 가이드 역할을 한다. - 노벨재단 제공
약물전달시스템 개발이 관건
아울러 많은 사람들이 RNA간섭의 임상 적용으로 관심을 돌렸다. 바이러스나 암의 유전자나 유전질환을 일으키는 변이 유전자를 표적으로 한 RNA간섭 약물이 나온다면 의학계에 혁명을 몰고 올 수 있기 때문이다.
1970년대 인트론(유전자에서 단백질의 아미노산으로 번역이 되지 않는 부분)을 발견해 1993년 노벨생리의학상을 받은 필립 샤프는 동료 과학자들과 투자자들을 끌어모아 2002년 바이오벤처 앨라이람을 설립했다. 이들은 RNA간섭의 잠재력을 높이 평가하고 연구에 뛰어들었는데 얼마 못 가 난관에 부딪혔다. RNA이중가닥을 표적이 되는 세포까지 보내는 게 생각보다 어려운 일이라는 사실이 드러났기 때문이다.
RNA는 설사 이중가닥일지라도 꽤 불안정한 물질이고 혈액에는 이를 인식해 파괴하는 효소가 있기 때문에 그대로는 약물로 쓸 수 없다. 바이러스 캡슐에 집어넣어(게놈 조각의 형태로) 표적이 되는 세포가 있는 조직을 감염시키는 방식도 문제가 많다.
결국 RNA이중가닥을 잘 감싸서 표적이 되는 조직에 갈 때까지 파괴되지 않게 만드는 약물전달시스템 개발에 RNA전달 약물의 미래가 걸려있다는 사실이 분명해졌다. 지난 10년 동안 앨라이람을 비롯해 RNA간섭 약물을 개발하는 벤처들은 숱한 시행착오를 거치며 이 연구를 해왔고 이번에 앨라이람이 마침내 RNA간섭 신약 승인을 받은 것도 제대로 작동하는 약물전달시스템를 개발한 덕분이다.
간단히 설명하면 RNA이중나선을 ‘지질나노입자(lipid nanoparticle)’ 안에 넣는 약물전달시스템이다. 즉 표면이 음전하인 RNA이중나선을 양전하인 지질 분자로 감싼 뒤 이를 다시 표면처리된(인체의 면역반응을 피하기 위해) 지질로 감싸 지름 50~100나노미터 크기의 나노입자로 만든 것이다.
RNA간섭 약물의 최대 걸림돌은 표적이 되는 세포까지 RNA이중가닥을 보내는 약물전달시스템을 찾는 일이다. 생체(혈액) 효소 시스템으로부터 RNA이중가닥을 보호해야 할 뿐 아니라 면역계의 표적이 되지 않아야 하고 세포에 흡수된 뒤에는 해체돼 안의 약물(RNA이중가닥)이 방출돼야 한다. 10여 년의 고투 끝에 찾아낸 유력한 약물전달시스템인 지질나노입자의 구조다. 이번에 승인을 받은 파티시란도 이 시스템을 이용했다. - ‘나노의학국제저널’ 제공
아직은 그림의 떡이지만
이번에 승인을 받은 RNA간섭 약물 파티시란은 변이 트랜스티레틴(transthyretin)의 mRNA를 파괴해 세포 내에 변이 트랜스티렌스 단백질이 축적되지 못하게 해 약효를 낸다. 이게 쌓이는 아밀로이드증(amyloidosis)은 세계에 환자 수가 5만여 명인 희귀병으로 발병하면 수년 내 사망한다. 즉 간에서 만들어진 변이 트랜스티레틴 단백질이 여러 조직에 축적돼 말초신경병증 등을 유발한다.
파티시란은 3주 간격으로 투약하는데 앨라이람이 제시한 약값으로 계산하면 1년 치료비가 무려 35만 달러(약 4억 원)에 이른다. 투약을 끊으면 다시 변이 단백질이 쌓이므로 결국 평생 투약해야 한다는 말인데 갑부가 아닌 다음에야 감당이 안 되는 수준이다(의료보험이 적용되면 달라지겠지만).
하지만 앞으로도 RNA간섭 약물이 이처럼 ‘그림의 떡’일 가능성은 낮다. 여러 업체에서 약물이 나오기 시작하고 제조시스템이 최적화되면 값은 빠르게 떨어질 것이다. 필자는 개인적으로 변이 유전자 질환 치료제보다는 항바이러스제나 항암제 쪽을 더 기대하고 있다.
이들이 변이를 일으켜 RNA간섭 약물에 내성을 갖게 돼도 그에 대응해 RNA의 염기서열만 바꿔주면 쉽게 극복할 수 있기 때문이다. 이는 단백질을 표적으로 하는 약물에서는 구현될 수 없는 이점이다.
유전성 트랜스티레틴 아밀로이드증 치료제인 파티시란의 작동 메커니즘을 도식화한 그림이다. 파티시란을 정맥투여하면(a) RNA이중나선이 들어있는 지질나노입자가 혈관을 타고 다니다 표적인 간세포로 들어간다(b. 나노입자 표면에 ApoE 단백질을 붙여 간세포 표면의 ApoE 수용체에 달라붙게 했다). 세포 안에서 나노입자가 해체되며(c) RNA이중가닥이 빠져나와 다이서에서 가공된 뒤 표적인 변이 트랜스티레틴(TTR) mRNA와 상보적인 단일가닥이 RICS에 결합돼 TTR mRNA를 파괴한다(d). 표적인 변이 트랜스티레틴(TTR) mRNA와 상보적인 단일가닥이 RICS에 결합돼 TTR mRNA를 파괴한다(d). 그 결과 변이 단백질이 축적되지 않아 아밀로이드증이 개선된다(e). ‘Pharmaceutical Research’ 제공
농약 내성 극복할 대안으로 떠올라
한편 앨라이람은 지난 2012년 거대 농약회사 몬산토와 10년짜리 협약을 맺어 RNA간섭 농약을 개발하고 있다. 오늘날 농약, 즉 살충제와 제초제는 내성이라는 위기를 맞고 있다. 즉 아무리 약을 쳐도 해충이나 잡초가 잘 죽지 않는다.
이에 대한 대응으로 해충의 유전자를 표적으로 한 RNA가닥을 만들어 살충제로 쓴다는 전략이 부상했다. 인간세포에 RNA조각을 넣는 일은 꽤 까다롭지만 절지동물의 경우는 RNA를 먹이면 장에서 쉽게 흡수되는 것으로 밝혀졌기 때문이다. 농작물에 농약을 치듯 RNA조각이 든 물을 뿌려주면 식물이 뿌리로 흡수하고 이를 먹은 해충이 RNA간섭으로 죽게 된다.
RNA간섭이 기대를 받는 이유는 특정 염기서열을 표적으로 하기 때문에 다른 생물체에 해를 끼칠 가능성이 낮기 때문이다. 또 해충이 내성을 획득할 경우 변이에 맞춰 RNA가닥을 새로 만들면 되므로 해충으로서는 죽을 맛이다.
앨라이람과 몬산토는 꿀벌의 해충인 진드기(학명 Verroa destructor)와 유채를 공격하는 벼룩잎벌레를 표적으로 하는 RNA간섭 살충제를 개발하고 있는데 2020년 시장에 나올 것으로 예상하고 있다. 한편 또 다른 거대 농약회사인 신젠타는 콜로라도감자잎벌레를 대상으로 RNA간섭 살충제를 개발하고 있는데 역시 2020년 출시를 목표로 하고 있다.
RNAi의 작용 메커니즘은 바이러스 공격을 사례로 들어 설명하 면 쉽게 이해할 수 있다. 바이러스가 세포내로 침입하게 되면 dicer라는 세포내 효소는 외부 침입물질인 바이러스의 dsRNA를 인지한 다음, 이를 21˜23 bp 정도 크기의 작은 조각으로 절단하 게 된다. 이 때 21˜23 bp의 크기로 잘려진 조각들을 흔히 small interfering RNA(siRNA)라 하는데, 세포내 단백질 복합체인 RNA induced gene silencing complexes(RISC)는 조각난 siRNA만을 특이하게 인지·결합하며, 결국 siRNA와 RISC의 결합체가 상보 적 염기서열을 갖는 바이러스의 mRNA를 선택적으로 찾아내어 분해를 유도하게 된다(그림 참조). 대부분의 동물세포에서는 dsRNA에 의해 RNAi가 일어나게 되 면, 인터페론 반응이 유도되어 모든 단백질 합성이 억제되고, 그 결과 아포프토시스(apoptosis)가 일어나 세포가 죽게 된다. 이런 까닭에 RNAi를 치료 목적으로 사용할 때 세포사멸 유도는 결정 적인 부작용이 될 것이라 우려가 있으나, 세포내로 짧은 RNA 조 각을 직접 주입하게 되면 세포사멸을 유도하지 않고 유전자 발현 만을 억제할 수 있을 것으로 기대되고 있다. 따라서 세포가 원천적으로 소유하고 있는 small RNA인 miRNA(micro RNA)에 대한 관심이 집중되고 있다. 기다란 머리 핀(long hairpin) 모양의 non-coding RNA가 dicer에 의해 절단·생성된 miRNA는 target mRNA에 불완전한 형태로 결합해 단백 질 합성을 억제하는데, 선충뿐만 아니라 사람을 비롯한 고등동물 에서도 이들의 정체가 확인되었다. 또한, 종에 따라 다소 차이는 있으나 그 수도 수백에서 수천 가지가 될 것으로 예상하고 있어 앞으로 각각의 기능과 작용 메커니즘을 규명하는 것이 miRNA를 치료제로 개발하는 과정에서 풀어야 할 과제이다.
RNAi의 활용방안
RNAi는 크게 신약개발을 위한 도구로서 이용되는 경우와 치료 제로 직접 개발되는 사례로 나눌 수 있다. 먼저 RNAi는 유전자의 기능을 규명하고 새로운 타깃 유전자를 발굴·확인하는 도구로 서 신약개발 과정 중 중요한 가치를 제공하고 있는데, 특히 게놈 프로젝트를 통해 밝혀진 수많은 유전자의 생체 내 기능을 밝히는 데 효율적으로 이용될 수 있는 기술이다. 지금까지는 주로 antisense, ribozyme, transgenic animal, 및 knock-out mouse 기 술 등을 이용해 유전자 기능을 분석해왔으나 대량분석이 어렵고 시간이 오래 걸리는 등 여러 가지 한계점과 단점이 보고 되어 있 어 새로운 기술이 요구되던 차였다. 특히 암과 같이 그 발생 기전 이 복잡한 많은 질환이나 또는 발생·분화·노화 등과 같이 정교 한 생명현상을 해석하기 위해서는 고속 및 대량분석이 가능한 RNAi가 가장 적합한 기술로 평가되고 있다. RNAi를 치료제로 개발하는 연구는 시장 잠재력이 매우 크기 때 문에 제약기업들의 관심이 집중된 분야이다. 하지만 적어도 10 년 이상의 연구가 더 진행되어야 임상 사용이 이루어질 것으로 예상되고 있다. 현재 in vitro 시험에서 종양발생과 바이러스 복 제과정에 결정적으로 관여하는 유전자를 침묵시키는 것으로 확 인됐고, 동물을 이용한 in vivo 시험에서도 RNAi가 autoimmune disease와 바이러스성 간염을 방어하다는 사실이 증명된 바 있 어, 주로 암, HIV 감염 및 신경퇴행성 질환을 치료 타깃으로 삼고 있다. 한편, RNAi의 개발기술은 ① RNAi 유도인자인 siRNA를 생성하 는 기술, ② siRNA를 세포내로 전달하는 기술, ③ siRNA에 의한 mRNA knockdown 확인 기술로 나눌 수 있다. 먼저, siRNA는 oligonucleotide 타입과 vector-based siRNA로 분류된다. 현재는 oligonucleotide siRNA가 80% 이상을 차지하 고 있는데, siRNA의 디자인 및 생성 과정에서는 siRNA가 실제 타 깃 유전자의 침묵을 유도하는 지 필히 확인해야 한다. 또한 siRNA는 세포내에서 작동하므로 이를 세포내로 전달하는 방법 의 개발이 핵심기술이 되고 있다. 기존의 transfection reagent외 에도 electroporation 기구의 사용, optoinjection 및 특정 펩타이 드를 이용해 siRNA의 전달능력을 향상시키는 방법 등이 새롭게 떠오르는 방법이다. 마지막으로는 siRNA에 의한 mRNA knockdown을 정량적 PCR 방법 등으로 확인하고, 무엇보다 western blot으로 단백질 합성이 억제되었음을 규명하는 기술이 필요하다. 이런 과정을 통해 이루어지고 있는 RNAi 개발연구는 이제 싹이 자라는 단계라 할 수 있다. 열매를 얻기까지는 아직도 많은 연구 가 필요한 분야라, 현재의 시장가치는 약 3천 4백만 달러에 지나 지 않고 있다. 그러나 전문기관에서는 매년 35% 이상의 성장을 지속하여 5년 후에는 약 1억 8천만 달러의 시장으로 성장할 것으 로 예상하고 있으며, 특히 선도 물질이 임상시험을 통해 가능성 을 보인다면 가히 폭발적인 시장 성장세를 보일 것으로 전망하고 있다.
RNAi : RNA Interference
Promega Note 83, p.33-36 By Natalie Betz. Ph.D., Promega Corporation
Abstract
RNA interference(이하 RNAi) 는 세포나 조직내에 특정한 mRNA target을 붕괴하여 인코팅 단백질을 넉-아웃시킴으로써 유전자 기능을 연구하는데 이용되는 매우 강력한 테크닉이며 급속히 전 파되고 있다. 이러한 특정 mRNA 붕괴는 상보적인 doublestranded RNA 가 매개한다. 이 기사는 RNAi 와 일반적으로 RNA siliencing 알려진 다른 RNA가 매개하는 기작과의 관계를 언급하 려 한다. 또한, 다양한 RNA siliencing 기작의 short interfering RNAs (이하 siRNAs), microRNAs (이하 miRNAs), small temporal RNAs (stRNAs), 그리고 short hairpin RNAs (shRNAs) 의 관계도 언급하였다.
History
RNAi는 dsRNA가 상보적인 타겟 mRNA의 특정한 분해를 자극해 서 타겟 단백질의 발현을 특이적으로 억제하는 현상이다 (참조 1-8 references. RNAi를 포함하여 일반적인 RNA silencing reviews). 이는 2001년 5월 RNA Society meeting에서 언급되었 으나, 이 현상에 대한 첫번째 힌트는 1990년 Rich Jorgensen 이 외부로부터 유전자를 도입하여 더 진한 보라색의 가공 페큐니아 를 만들고자 시도한 것에서 예기치 않게 다양화된 색소형성을 보 여줌으로써 알려졌다(9). 도입된 DNA sequence 는 endogenous loci의 발현에 어느 정도 영향을 미치는데, 이 현상을“cosuppression”이라 명명하였다. 동시에, 식물 실험들에 의해 식물 들이 분해를 하도록 특별히 설정된 viral RNA로 인한 RNA 바이 러스 감염에 반응하는 것이 알려졌다(10,13). 식물에서 관찰되는 이러한 현상들을“post-transcriptional gene silencing”(이하 PTGS), 또는“viral-induced gene silencing”(이하 VIGS) 라 명명 하였다(14,15). 1995년 Guo 와 Kemphues 는 C. elegans 에서 sense RNA 가 antisens RNA 만큼이나 유전자 발현을 억제하는 효과가 있음을 발견하였다(16). 이 연구는 Fire et al. 등에 의해 seminal RNAi 논 문에까지 확대되었으며, 여기서 dsRNA 가 C. elegans 에서 sense 나 antisense RNAs 각 각과 비교해 10배 이상 유전자 발현을 감 소시키는 것을 서술하였다(17). C. elegans 에서의 연구 이후에 RNAi는 여러 유기체들, 예를 들 면 관상어(18), 플라나리아(19), 히드라(20), 진균류(21), 초파리 (22), 그리고 마우스의 embryo(23,24)를 대상으로 한 실험에서도 입증되고 있다. Neurospora crassa에서처럼 곰팡이에서도 dsRNA 에 대한 반응을“quelling”이라 불렀는데, 이것을 그 유전 자의 형질전환 카피의 도입으로 유발된 endogenous gene의 침 묵(silencing)이라 불렀다. 이러한 현상들을 모아 RNA silencing 이라 명명하였고, 일련의 단백질과 짧은 RNAs를 이용하는 것으로 알려지고 있다. 이러한 과정들은 동일하진 않아도 기작적으로 매우 유사하다.
Mechanism
대부분의 포유류 시스템에서, 매우 긴 dsRNAs (> 30bp) 의 도입 은 dsRNA-activated protein kinase, PKR (EIF-2α를 인산화하는) 을 활성화하여 일반적인 translation 억제를 유발시키는 잠재적인 antiviral 반응을 야기한다. 게다가, ds RNA는 2’, 5’ oligoadenylate polymerase/RNase L system을 활성화시키고 apoptosis를 통해 세포사를 유도할 수 있는 IkB를 억제시킨다. 따라서, Tuschl/Fire과 동료들이 화학적으로 합성된 siRNAs가 apoptosis를 야기하지 않고도 다양한 mammalian cell lines에서 특이적인 유전자 silencing을 유도할 수 있다는 것을 보여준 것은 매우 반가운 소식이다 (26,27).이것은 또한 Drosophila embryo extracts (in vitro)에서도 관찰되었다 (28-31). 기작적으로, RNAi는 여러 다양한 과정에 영향을 미친다 (Figure 1). dsRNA는 RNase Ⅲ family member (e.g., Dicer in Drosophila)에 의해 인식되어 21-23 nucleotides의 siRNAs로 절 단된다 (28,32,33). 다음으로, siRNAs는 RNAi targeting complex 로 알려진 RISC (RNA-induced silencing complex)와 결합하여 완전한 siRNA와 일치하는 mRNAs를 절단한다 (32,33). 타겟 mRNAs는 siRNA (28)와 상보적인 부분의 중앙에서 절단되어, 타 겟 mRNA의 급격한 분해를 초래하여 단백질 발현이 감소된다. 가장 강력한 siRNA duplexes는 각 각의 말단에 2-uridine을 가지 18 www.seoulin.co.kr RNAi introduction 고 19bp 서열로 구성된 21 nucleotide이다 (28). Human에 일치하는 Dicer가 클로닝 하여 발현시켰고 더 나아가 특성을 밝혔다. 그리고 소포체에서 calreticulin과 함께 존재하는 것이 밝혀졌다 (34,35). Dicer는 DexH/DEAH RNA helicase/ATPase domain, PAZ signature, 2개의 neighboring RNase Ⅲ-like domains, 그리고 dsRNA binding domain (RBD)을 포함하는 거대한 multidomain 효소이다. Dicer가 dsRNA 절단할 때는 ATP를 필요로 하지 않지만, 아마도 잘린 product release에 는 ATP가 필요할 것이다 (34). Human cells에서 RNAi는 세포질 또는 핵의 밖으로 나가는 mRANs에서만 보이는 것으로 알려졌다 (36). 여러 다양한 진핵 생물들의 RNAi 사이에서 두드러진 차이점은 Drosophila와 mammals에서처럼 자발적이고 유전되지 않는 silencing 과 C. elegans에서처럼 silencing이 유전될 수 있고 체 계적인 형태를 보인다는 것이다 (7). 유사하게 체계적인 silencing은 식물에서 관찰되었고, 최근 확인된“transitive RNAi” 라는 현상은 C. elegans와 더 진화된 진핵 생물 사이에서의 차이 점을 파악할 수 있게 할지도 모른다. Transitive RNA는 특이한 유전자를 통한 RNAi silencing signal의 이동을 의미한다. 예를 들어, C. elegans에서 mRNA의 3’- position 타겟팅은 타겟 부위와 유사한 siRNAs 생성과 그것의 전 사 억제를 초래한다. 게다가, silencing trigger에 의해 처음에 타 겟된 mRNA upstream의 위치에 상보적인 siRNAs는 또한 축적되 는 것으로 보인다. 만약 이 siRNAs가 다른 mRNAs와도 상보적이 라면, 그 mRNAs는 아주 잘 silencing에 타겟이 될 것이다. 따라 서, silencing은 특이적인 mRNA target에서 3’에서 5’로 진행될 수 있고, 최초의 target에서 서열 유사성을 갖는 부위를 공유하는 다른 mRAN targets에서도 진행될 수 있다. Transitive RNA 현상은 RNA-의존적인 RNA polymerase (RdRP) 가 필요하고, RdRPs에 의해 dsRNA (그리고 siRNAs)가 생성되는 기본적인 합성을 준비하는 모델로 유도하는 것으로 보인다 (37). 그러나, RdRPs는 날벌레나 포유류의 RNAi에 필수적인 것은 아 니다 (38,39).
Synthesis and Use of dsRNA
긴 double-stranded RNAs (~200-1,000bp)나 짧은 siRNAs (21- 23bp)의 합성은 T7 RNA polymerase와 함께 전사(in virto)에 사 용됨으로써 완성된다 (17,40,41). siRNAs는 화학적으로 합성되어 질수 있고, 가장 널리 사용되는 Dharmacon Research, Inc. 에서 공급하는 siRNA같이 상업적인 판매가 많이 이루어지고 있다. Yu et al. (41)은 in vitro 상에서 합성된 siRNA와 화학적으로 합성된 siRNA사이에서 유사한 효율을 입증하였다. 더욱이, siRNAs는 정 제된 E.coli Exonuclease Ⅲ와 함께 in vitro 상에서 긴 dsRNAs로 부터 digestion을 통해 만들어질 수 있다 (42,43). 효소적으로 긴 dsRNAs가 siRNAs로 잘리는 과정은 단백질 수준에서의 감소와 감지할 수 있는 생체 반응의 가능성을 증가시켜 주면서, 타겟 mRNA에서 여러 사이트와 상호작용을 할 수 있는 다양한 siRNAs 의 생성으로 연결된다. 게다가 최근에, in vivo에서 특이하게 디자인된 expression vectors의 사용은 세포와 유기체에서 dsRNA/siRNA의 합성과 도 입을 미연에 방지한다. 잘 정의된 전사 개시 시그날과 5개의 연 속적인 thymidine 잔기로 구성된 종결 시그날을 갖고, 발생기의 RNA 전사의 절단은 두번째 uridine (44) 다음에 발생하는 RNA polymerase Ⅲ promoters를 이용하여 in vivo에서 적합한 구조로 siRNAs 합성이 일어나게 한다. HI (45,46)와 U6 (41,47) promoters는 이 목적으로 사용되고 있다. 게다가 viral 또는 retroviral 벡터의 사용하여 세포나 조직내에서 siRNAs의 합성(in vivo)이 성공적임을 보여주고 있다 (48,49). In vivo에서 siRNAs 의 발현은 일반적으로 shRNAs를 형성하는데, 이 형성은 완전하 게 일치하는 base pairing의 19개의 nucleotide가 2-nucleotide 3’ www.seoulin.co.kr 19 BIOTIMES Figure 1. Proposed mechanism of RNAi. RNase III family에 속하는 dsRNA-processing 단백질은 dsRNA와 결합한다. RNase III 는 dsRNA를 siRNA로 절단한다. siRNA는 multicomponent nuclease complexes (RISC)를 형성한다고 제안되고 있다. RISC에 의해 인식되는 타겟 mRNA는 siRNA에 상보적인 부분의 중앙이 절단되고 타겟 RNAs는 급속히 분해 된다. overhang 부분에서 다양한 spacer regions과 끝부분에 의해 연결 되는 것으로 형성되는 것이다 (41,45,49). McManus et al. (47)은 hairpin 구조에서 약간의 변형이 silencing activity에 영향을 미치 므로 완전한 전사의 디자인이 매우 중요함을 입증하였다. RNAi 개시를 위한 dsRNA/siRNA의 도입은 수많은 기술을 사용 함으로써 완성될 것이다. C.elegans의 경우, dsRNA는 주입되거 나(17,50), 스며들거나(51), 흘러들어갈 수 있다(52-54). Drosophila 세포는 배양배지에서 dsRNA에 쉽게 노출되는 반면 에(55), mammalian 세포에서 siRNA의 도입하기 위해서는 transfection 시약과 방법이 필요하다 (26,27).
RNA Silencing and Other Small RNAs
비록 miRNAs라 불리는 수많은 작고 단일 stranded RNAs가 확인 되었다 하더라도, 단지 lin-4와 let-7 miRNAs 2가지의 기능이 알 려져 있다. 이것은 C.elegans에서 일시적으로 발현되므로 stRNAs라 불린다 (56). Let-7은 (모두 그런 것은 아니겠지만) 초 파리와 인간 같은 좌우대칭인 동물 대부분에 존재한다 (57). 작 고 일시적인 RNAs는 stRANs와 상보적인 3’-untranslated regins 을 포함하는 mRNAs에 의해 암호화된 단백질 발현을 반대로 조 절함으로써 진보적인 진행을 유도한다 (56). 이 과정을“번역 억 제”라 부르는데 hybridized mRNA와 완전한 상보적일 필요는 없 다 (58). Dicer와 그 유사한 것들로 인해 안정하고 큰 stRNAs (~70 nucleotide)가 stemloop recursors 진행되는 것은, siRNAs와 stRNAs 생성하는데 필요한 일반적인 기작을 보여주는 것이다 (59-61). 미세한 siRNAs와 miRNAs인 합성된 헤어핀 RNAs는 mRNA 분해를 통하여 silencing 하는 유전자를 효율적으로 타겟 할 수 있다 (47). RISC는 다른 조절 RNA 종들이 보충되어 다른 RNA silencing 메커니즘을 초래하는 유동성이 있는 플래트폼이 다 (7). 그러나, 타겟 단백질 양의 감소는 항상 나타난다.
RNAi and the Genome
최근 연구는 RNAi를 매개하는 단백질 성분들과 short RNAs는 RNAi를 연결시키고 유전자적인 완전함을 유지하는 필수적인 chromosomal 기능이 반드시 필요하다는 것을 보여주고 있다. RNA-based silencing 메카니즘은 바이러스와 전이될 수 있는 인 자들에 대항하여 스스로 보호하고 불완전한 mRNAs를 제거하는 면역체계의 전구체로써 작용할지도 모른다. RNAi 기법은 식물에서 genomic methylation을 유도하고 초파리, C.elegans 그리고 진균류에서 chromatin 구조를 변형시킨다 (7). Applications and Future Directions RNAi는 mammalian cell에서 세포 성장에 필수적인 gene products를 밝히기 위하여 사용되고 있다 (62). 게다가, Irie et al. 은 human과 rat 세포 모두에서 다양한 PKC isoform의 sub-type 과 species-specific knockdown을 기술하였고, thymine dimmer antibody를 이용하여 UV-irradiated siRNA-transfect한 세포를 탐 색하였다 (63). RNAi는 종양 유전자 생산물의 분해를 타겟으로 사용되는데, 이 것은 이 단백질에 의해 형성된 백혈병 세포들의 apoptosis에 의 해 순차적인 죽음을 초래한다 (64). 세포배양이나 동물들에서 HIV-1(65)나 HCV(66)에 의해서 특이적으로 타겟되고 바이러스 감염을 예방하는 RNAi의 사용은 잘 서술되고 있다. Poliovirus (소아마비 병원체) 감염은 capsidprotein mRNA나 viral polymerase gene을 타겟으로 하는 siRNAs를 감소시킨다 (67). Xia et al.에 의한 연구는 마침내 polyglutamine 질병을 타겟으로 하는 in vitro와 in vivo에서 바이러스에 의해 매개되어지는 delivery methods의 사용을 이루어냈다 (48). 이 발견은 RNAi와 siRNAs 또는 shRNAs를 사용하여 새로운 유전자 치료가 항암 또 는 항바이러스성 치료로 이어지도록 하는 분야를 열어 주었다 (68).
Conclusions
내생적인 발현 또는 외부로부터 도입된 short RNA 분자에 의해 조절되는 RNA interference와 다른 RNA silencing 현상은 오늘날 단일 세포인 원생동물에서 포유동물에 이르는 생물학의 진화를 가져오고 있다. Mammalian cells에서 loss-of-function genetic screens과 유전자 상호작용 테스트가 빠르게 이루어질 것이나 그 러기 위해서는 많은 어려움이 예상된다(7)